
Contents
LHON disease
LHON is short for Leber Hereditary Optic Neuropathy also called Leber’s disease, Leber’s optic atrophy or Leber’s optic neuropathy, is a rare inherited eye condition that causes sudden, painless and irreversible loss of central vision 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11. Leber hereditary optic neuropathy (LHON) affects the optic nerve in your eye. The optic nerve is the second cranial nerve (CN II) that is comprised of about 1.2 million of nerve fibers called retinal ganglion cells that send visual messages to your brain to help you see 12. You have an optic nerve at the back of each eye that connects directly to your brain. Each optic nerve is like the electric cable that carries electrical signals in a one-way connection, and it only carries signals from your eyes to your brain 12. Some of the signals in the optic nerve also contribute to other abilities and brain processes. Leber hereditary optic neuropathy (LHON) usually begins as a one sided (unilateral) progressive optic nerve damage (optic neuropathy), with sequential involvement of your other eye months or years later 13. Symptoms typically begin in young adulthood and include blurred or cloudy vision, making reading, driving, and recognizing faces difficult, with vision loss often affecting one eye before the other. If vision is lost, then it usually occurs before 40 years of age.
The onset of Leber hereditary optic neuropathy (LHON) is acute, with patients developing blurred central vision very quickly. Leber hereditary optic neuropathy (LHON) is often characterized by painless subacute loss of central vision that may begin in one eye or both eyes simultaneously during young adult life 1. In most cases, symptoms begin with one eye first, followed a few weeks later (on average eight weeks later) by visual failure in the other eye. Over time, the vision in both eyes typically worsens, affecting central and color vision. In a small percentage of cases, the central vision loss can improve; but in most cases loss of vision is permanent. The severity of symptoms may vary from one affected individual to another, even within the same family, due to a ‘dosage’ effect. This is due to the fact that you have many mitochondria in each cell. In one individual, if only a small proportion of mitochondria in each cell have the mutation, symptoms will be mild. In another individual, if a higher proportion of mitochondria in each cell carry the mutation, symptoms will be more severe.
The standard version of LHON (Leber hereditary optic neuropathy) only affects your optic nerves, and vision loss is its only symptom. This is the case for the vast majority of people with Leber’s disease. But in rare cases, some people with LHON have additional symptoms that affect other parts of their nervous system such as peripheral neuropathy, postural tremor, nonspecific myopathy (a broad term for a muscle disorder that involves muscle weakness, fatigue, or stiffness that doesn’t fit into a specific category and isn’t caused by inflammation), dystonia (a movement disorder that causes muscular spasm and abnormal posture), and multiple sclerosis–like illnesses and sometimes heart conduction abnormalities. Doctors call this version of LHON disease “Leber plus” or “LHON Plus” disease.
People with “Leber’s plus” or “LHON Plus” disease can have a variety of symptoms beyond vision loss, including 14:
- Movement disorders or motor disorder, like tremors.
- Coordination and balance problems (ataxia).
- Spasticity
- Cardiac conduction problems, like arrhythmias (irregular heartbeats).
- Multiple sclerosis-like syndrome such as muscle weakness and fatigue.
- Postural tremor.
- Spinal cord disease.
- Skeletal changes.
- Parkinsonism with dystonia.
- Anarthria – a severe motor speech disorder characterized by the complete inability to speak due to a lack of muscle control for articulation.
- Motor and sensory peripheral neuropathy.
- Mild encephalopathy.
- Cardiac arrhythmias.
LHON is caused by genetic mutations in the mitochondrial DNA (mtDNA), the “powerhouses” of your cell, which is inherited solely from your mother 11. You inherit all of your mitochondria from your birth mother, so only female parents can pass the gene mutation to their biological children. But not everyone who carries the mutation develops symptoms of LHON, so not everyone realizes they carry it. Mothers with a LHON gene mutation may not show symptoms, but family history often reveals maternal relatives with visual loss at an early age. Mutations in mitochondrial DNA (mtDNA) can only be inherited maternally because mitochondria derive from ova, not sperm. All of the offspring of a mother with a mitochondrial DNA (mtDNA) mutation will inherit the gene. However, it is currently impossible to predict which members of a family who carry the mitochondrial DNA (mtDNA) mutation will eventually develop vision loss. Often, people who develop LHON have no family history of the condition. The prevalence of visual loss from LHON is approximately 1 in 25,000 to 50,000 people. For unknown reason, approximately 80% to 90% of people with LHON are male. A male with a mitochondrial DNA (mtDNA) mutation cannot transmit the mutated gene to any of his children. Most carriers never suffer significant visual loss; males are about four to five times more likely than females to lose vision and be affected. The incidence of visual loss, therefore, is much less and about 1:10 million/year.
LHON is classically associated with mitochondrial base pair mutations G11778A (guanine to adenine at position 11778), T14484C (tyrosine to cytosine at position 14484), and G3460A (guanine to adenine at position 3460) also known as mitochondrial LHON (mLHON), which account for over 90% of LHON patients 10, 15, 16, 17, 18, 19, 20, 2. These mutations primarily affect respiratory chain complex I genes and the mitochondrial genes ND1, ND4, and ND6, among others 2. The most common LHON-causing mutation is G11778A (guanine to adenine at position 11778) mutation, reported to account for 70% of LHON cases in Northern European populations and 90% of LHON cases in Asian populations 2. The G11778A (guanine to adenine at position 11778) mutation is associated with more severe disease, with less chance of significant visual recovery. The T14484C (tyrosine to cytosine at position 14484) mutation is associated with the best overall prognosis, with some visual recovery in 37% to 58% of LHON cases 2. This typically occurs after reaching a visual “nadir” (lowest visual acuity). The T14484C (tyrosine to cytosine at position 14484) mutation has been seen in French Canadian populations with LHON 21, 22. The G3460A (guanine to adenine at position 3460) mutation usually portends a more intermediate course and has the worst prognosis 21, 22.
Recessive forms of LHON also known as autosomal recessive LHON (arLHON) have recently been described, caused by mutations in the DNAJC30, MCAT, MECR, and NDUFS2 genes 23, 24, 25, 26, 27.
While LHON is genetic, risk factors that can trigger or worsen the optic neuropathy include smoking, alcohol use, environmental toxins, systemic illness, and psychological stress. To potentially reduce your risk or severity of progressive optic nerve damage (optic neuropathy), avoid smoking, alcohol consumption and exposure to environmental toxins.
LHON is diagnosed based on ophthalmologic findings, which include specialized visual testing. Your eye specialist (ophthalmologist) will conduct standard eye exams to test your vision and look for the cause of your problem. Ophthalmologic testing involves dilated fundus examination to identify characteristic changes in your optic disc and vascular changes during the acute phase, visual fields, electrophysiologic studies and imaging, particularly Optical Coherence Tomography (OCT, an eye test is a non-invasive imaging test that uses light waves to take high-resolution, cross-sectional pictures of the retina and optic nerve). Your ophthalmologist might not be able to see anything wrong with your eye or your optic nerve at first. The damage becomes more obvious after the first six months of symptoms. When your ophthalmologist suspects LHON, he/she will recommend genetic testing for mitochondrial genes associated with LHON to confirm that you have one of the gene mutations involved. Most affected individuals know if their family members also are affected by LHON.
There is no cure for LHON, but treatments focus on managing the condition through support, adaptive technologies, and aids. Clinical trials are looking at the use of antioxidant supplements and gene therapy. Coenzyme Q10, L-carnitine, creatine, lipoic acid, lutein, dimethylglycine, cysteine, succinate, dichloroacetate, vitamin K1, vitamin K3, vitamin C, vitamin B1, vitamin B2, and vitamin E have all been suggested as treatment for LHON, but there is currently insufficient evidence to support their use 28, 29, 30. Several studies have shown that therapies involving ubiquinone and idebenone may provide possible benefits during both the acute and chronic phases of the disorder 31, 32. Idebenone is a synthetic short-chain benzoquinone thought to restore mitochondrial function by bypassing the dysfunctional complex I and thus restoring mitochondrial adenosine triphosphate (ATP) generation and by acting as a potent antioxidant 33, 34, 35, 36. Recently, additional modes of action have been proposed, including effects on apoptosis, mitophagy, and myelination 37, 38.
In the randomized, double-blind, placebo-controlled Rescue of Hereditary Optic Disease Outpatient Study (RHODOS), patients with LHON and disease onset ≤5 years were treated with idebenone (300 mg 3 times/day) or placebo for 6 months 39, 21. A trend toward improved visual acuity (VA) was observed in idebenone-treated patients. In hindsight, the 6-month treatment duration was likely too short to fully capture the potential treatment benefit. An expanded access program allowed for analysis of long-term idebenone treatment in the real world, in subacute/dynamic LHON patients (≤1 year after onset) 40. This noncontrolled study indicated the potential benefit of maintaining idebenone therapy for 24 to 30 months before classifying patients as nonresponders. This approach resulted in a visual acuity (VA) stabilization and/or recovery rate that was higher than expected from limited natural history studies 41, 42. Based on this cumulative clinical evidence, the European Medicines Agency approved idebenone (Raxone) for the treatment of individuals ≥12 years old with LHON 43. Raxone (idebenone) can only be obtained with a prescription and treatment should be started and supervised by a doctor with experience in LHON. Raxone (idebenone) is available as 150 mg tablets, and the recommended dose is two tablets taken three times a day with food 43. Long-term efficacy studies for idebenone in LHON are limited by the lack of direct control data, which are difficult to prospectively compile for rare diseases with an approved treatment. In addition, little data have been collected in the chronic phase (>1 year after onset) 44, 45.
An open-label, international, multicenter, natural history-controlled LEROS study assesses the efficacy and safety of idebenone treatment (900 mg/day) in patients with LHON up to 5 years after symptom onset (N = 199) and over a treatment period of 24 months, compared to an external natural history control cohort (N = 372), matched by time since symptom onset 46, 47. The LEROS study meets its primary endpoint based on clinically relevant benefit at month 12 and confirms the long-term efficacy of idebenone in the subacute/dynamic and chronic phases; the treatment effect varies depending on disease phase and the causative mtDNA mutation. LEROS confirmed the benefit of idebenone in LHON, including in the chronic phase (1–5 years since onset) with a consistent treatment benefit observed for patients with the most common G11778A (guanine to adenine at position 11778) mutation regardless of disease phase, and for patients with the T14484C (tyrosine to cytosine at position 14484) mutation in the chronic phase. LEROS confirms a favorable safety profile of idebenone in LHON patients. Howver, further study of idebenone use in patients carrying the G3460A (guanine to adenine at position 3460) mutation is needed to clarify treatment benefits. In the meantime, patients carrying G3460A mtDNA mutation who are in the subacute/dynamic phase should be adequately counseled to allow them to make an informed decision as to whether treatment with idebenone should be initiated. The findings of the LEROS study will help guide the clinical management of patients with LHON 47.
A second drug, the antioxidant alpha-tocotrienol-quinone (EPI-743), a vitamin E derivative, has also shown promising early results and may have more potency than idebenone. A small open-label study of five patients with acute LHON showed that early treatment with EPI-743 arrested disease progression in four out of five the LHON-patients 48, 49. However, a double-blind, randomized placebo-controlled trial is needed to provide confirmation of EPI-743 in the treatment of acute LHON.
Currently the most exciting potential means of treating LHON are within the field of gene therapy 6, 50, 51. There are several forms of gene therapy which are being evaluated as possible treatments for LHON, at various stages of testing.
If you or a family member have a family history of LHON, consider seeking genetic counseling for support and future planning. Genetic counseling is recommended for affected individuals and maternal family members. Male patients may be reassured that they cannot transmit the disease to their offspring. The mother of an affected individual likely harbors the mitochondrial DNA mutation, but may or may not develop symptoms. Female patients will transmit the mutation to all offspring. However, those who exhibit heteroplasmy may transmit a low mutation load; therefore, offspring may have a low risk of disease. De novo mutations are rare but may be present in individuals whose maternal relatives do not have a history of visual loss.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Anatomy of the eye
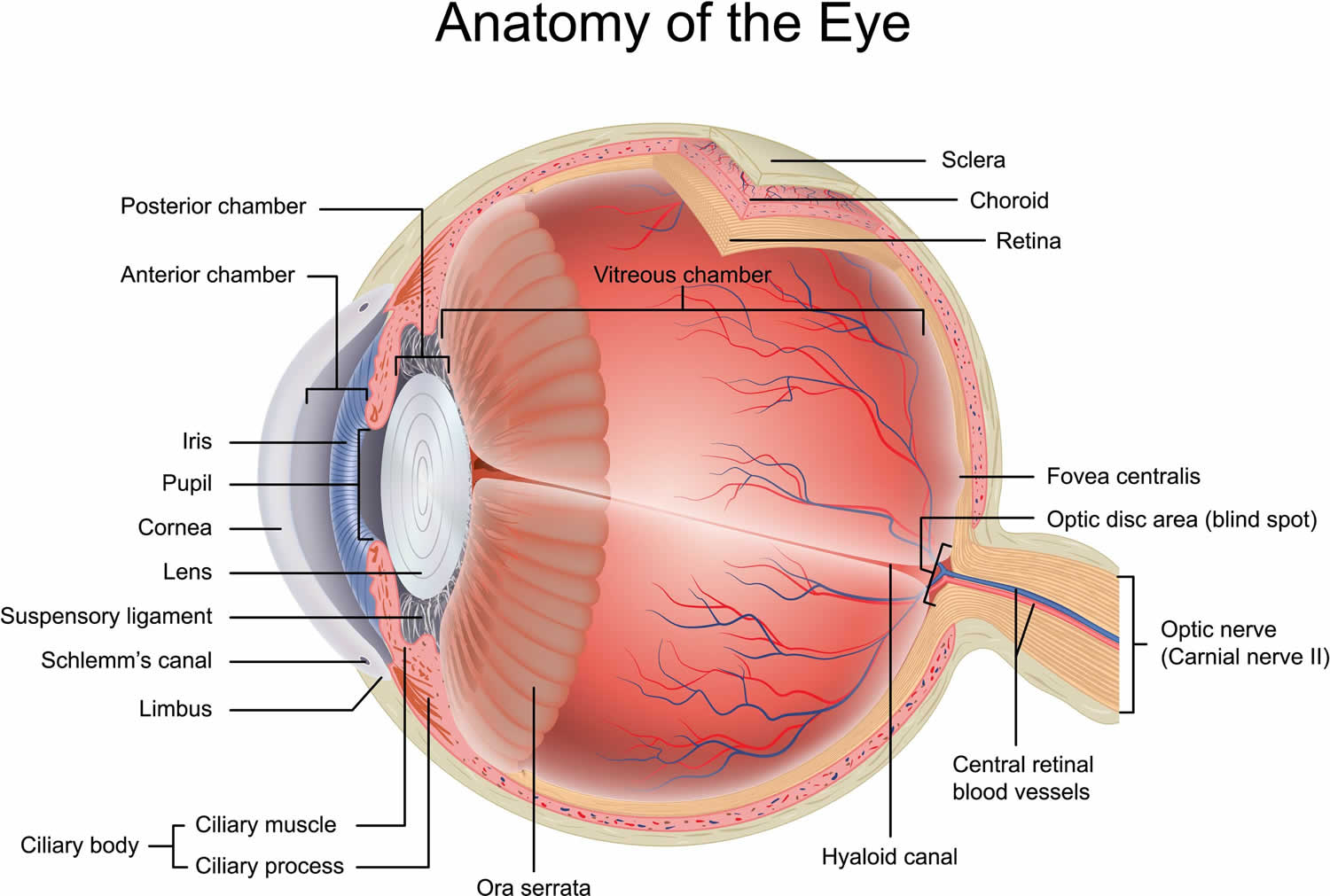
Figure 2. Leber Hereditary Optic Neuropathy
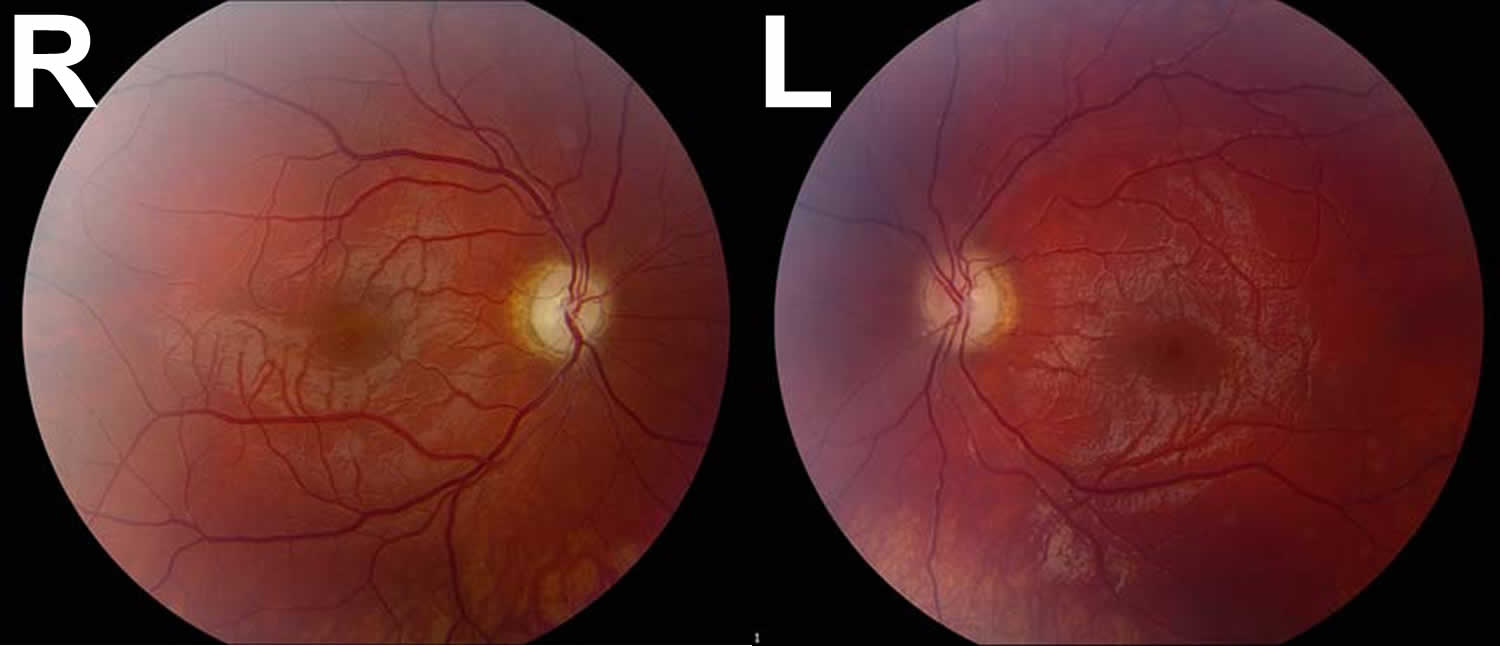
Footnotes: A 17-year-old male was referred to ophthalmology clinic by his local optometrist for evaluation of severe, painless, bilateral vision loss. Four months prior to presentation, the patient had normal uncorrected vision and passed his driver’s license vision screening. He first noted vision difficulties when trying to follow the flight of a golf ball two months prior to presentation and later had trouble with reading. The patient could not recall a specific moment of worsening and he was unsure whether one eye was affected prior to the other or if there was synchronous involvement. Dilated fundus examination revealed the development of temporal optic nerve pallor with loss of hyperemia both eyes. Despite an unclear history of sequential vision loss between the two eyes and a lack of family history for visual loss, the appearance of the optic nerves, painless bilateral vision loss, young age, and male gender was suspicious for Leber hereditary optic neuropathy (LHON). His genetic testing came back positive for the 11778 mitochondrial DNA point mutation confirming the diagnosis of LHON.
[Source 52 ]Figure 3. Leber Hereditary Optic Neuropathy OCT
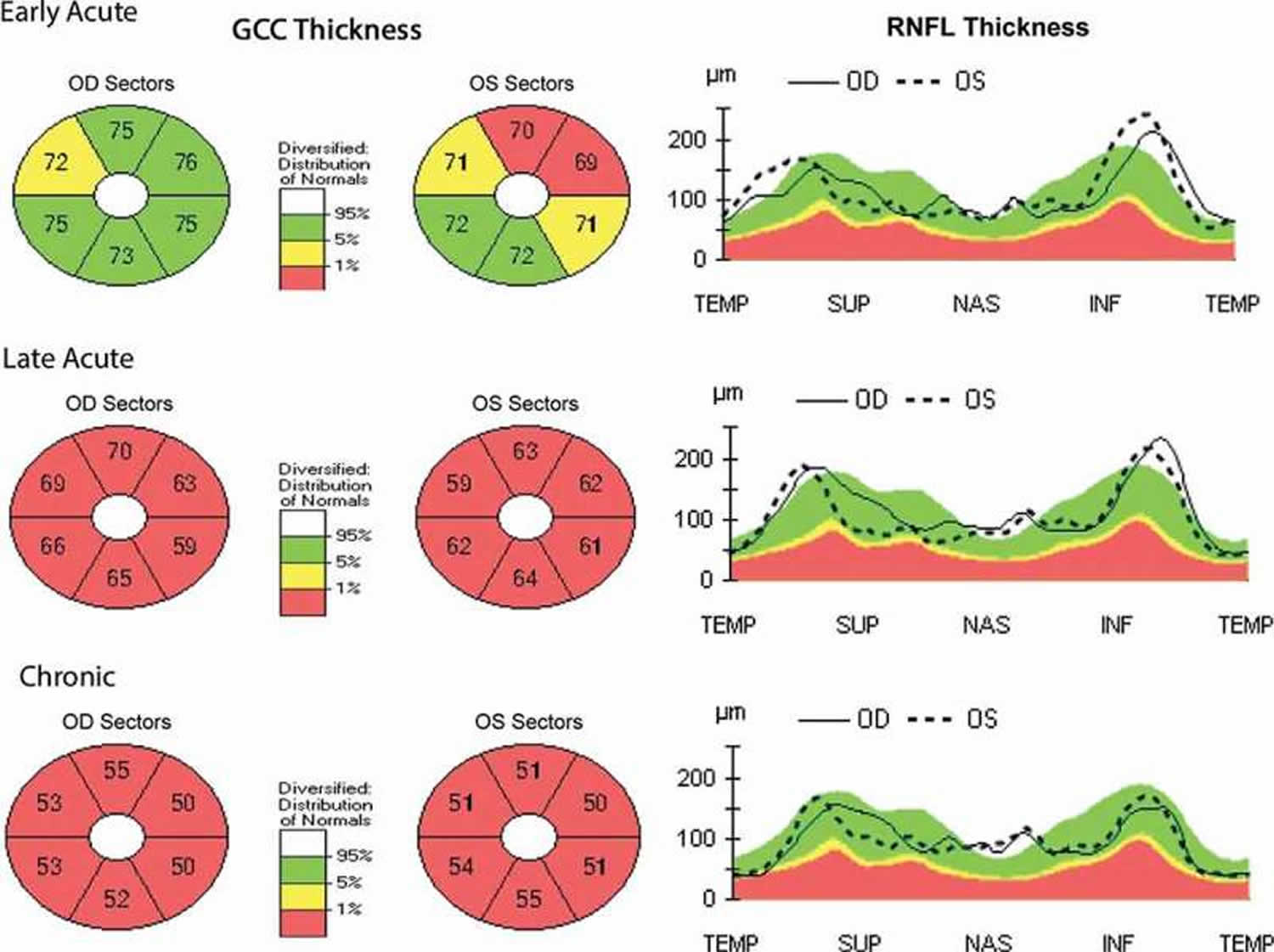
Footnotes: Retinal Nerve Fiber Layer (RNFL) is a layer of nerve fibers on the inner surface of the retina that transmits visual information to the brain. The ganglion cell complex (GCC) is a group of three innermost layers of the retina, the nerve fiber layer, the ganglion cell layer, and the inner plexiform layer, that collectively process and transmit visual information from the eye to the brain. Analysis of the RNFL and GCC is crucial for assessing optic nerve health, as a thinner layer or other defects can indicate diseases like glaucoma, which damage the optic nerve. Optical coherence tomography (OCT) findings throughout the early acute, late acute, and chronic phases in LHON patient with decreased vision bilaterally (20/30 and 20/50) 3 weeks after symptom onset. In the early acute phase, retinal ganglion cell complex (GCC) defects are noted bilaterally with RNFL swelling. In the late acute phase, severe retinal ganglion cell complex (GCC) thinning is present with persistent RNFL swelling. In the chronic phase, severe GCC thinning is present with RNFL thinning noted temporally.
Abbreviations: OD (Oculus Dexter) = Right Eye; OS (Oculus Sinister) = Left Eye
[Source 53 ]LHON disease causes
LHON is caused by genetic mutations in the mitochondrial DNA (mtDNA), the “powerhouses” of your cell, which is inherited solely from your mother 11. You inherit all of your mitochondria from your birth mother, so only female parents can pass the gene mutation to their biological children. But not everyone who carries the mutation develops symptoms of LHON, so not everyone realizes they carry it. Mothers with a LHON gene mutation may not show symptoms, but family history often reveals maternal relatives with visual loss at an early age. Mutations in mitochondrial DNA (mtDNA) can only be inherited maternally because mitochondria derive from ova, not sperm. All of the offspring of a mother with a mitochondrial DNA (mtDNA) mutation will inherit the gene. The prevalence of visual loss from LHON is approximately 1 in 25,000 to 50,000 people. For unknown reason, approximately 80% to 90% of people with LHON are male. A male with a mitochondrial DNA (mtDNA) mutation cannot transmit the mutated gene to any of his children. Most carriers never suffer significant visual loss; males are about four to five times more likely than females to lose vision and be affected.
LHON is classically associated with mitochondrial base pair mutations G11778A (guanine to adenine at position 11778), T14484C (tyrosine to cytosine at position 14484), and G3460A (guanine to adenine at position 3460) also known as mitochondrial LHON (mLHON), which account for over 90% of LHON patients 10, 15, 16, 17, 18, 19, 20, 2. These mutations primarily affect respiratory chain complex I genes and the mitochondrial genes ND1, ND4, and ND6, among others 2. The most common LHON-causing mutation is G11778A (guanine to adenine at position 11778) mutation, reported to account for 70% of LHON cases in Northern European populations and 90% of LHON cases in Asian populations 2. The G11778A (guanine to adenine at position 11778) mutation is associated with more severe disease, with less chance of significant visual recovery. The T14484C (tyrosine to cytosine at position 14484) mutation is associated with the best overall prognosis, with some visual recovery in 37% to 58% of LHON cases 2. This typically occurs after reaching a visual “nadir” (lowest visual acuity). The T14484C (tyrosine to cytosine at position 14484) mutation has been seen in French Canadian populations with LHON 21, 22. The G3460A (guanine to adenine at position 3460) mutation usually portends a more intermediate course and has the worst prognosis 21, 22.
Recessive forms of LHON also known as autosomal recessive LHON (arLHON) have recently been described, caused by mutations in the DNAJC30, MCAT, MECR, and NDUFS2 genes 23, 24, 25, 26, 27.
A significant percentage of people with a mutation that causes LHON do not develop any features of the disorder. Specifically, more than 50 percent of males with a mutation and more than 85 percent of females with a mutation never experience vision loss or related health problems. Additional factors or risk facotrs may determine whether a person develops the signs and symptoms of LHON. Environmental factors such as smoking, alcohol use, environmental toxins, systemic illness, and psychological stress may be involved, although studies have produced conflicting results. Researchers are also investigating whether changes in additional genes contribute to the development of signs and symptoms. To reduce your risk or severity of progressive optic nerve damage (optic neuropathy), avoid smoking, alcohol consumption and exposure to environmental toxins.
Many mitochondrial diseases display heteroplasmy. Heteroplasmy is the presence of more than one type of mitochondrial DNA (mtDNA) sequence within a single cell or individual 54. This mixture of both wild-type (normal) and mutated (variant) mitochondrial DNA (mtDNA) is a normal genetic variation, though it can be clinically significant if the proportion of mutated sequences becomes high enough to cause a mitochondrial disorder. It can arise through inheritance from one or both parents or through mutations that occur during a person’s lifetime. Each cell contains numerous mitochondria, and in 10% to 15% of patients with LHON mitochondrial mutations only a certain percentage of these mitochondria may have mutant DNA. Those with a “mutation load” less than a certain threshold (60%–75%) may not exhibit LHON disease clinical features and may never lose vision 2. It has been theorized that due to the heteroplasmy seen in LHON, the right and left eyes may have different amounts of affected mitochondrial DNA (mtDNA) 55.
The mitochondrial DNA (mtDNA) mutations cause defects in several NADH-ubiquinone oxidoreductase chains 2, 11. This is thought to impair glutamate transport and increase reactive oxygen species production in retinal ganglion cells via apoptosis (programmed cell death), though it is unclear why these cells are particularly vulnerable. Atrophy and demyelination are subsequently noted in the optic nerves, chiasm, and tracts 2.
In acute LHON, macular changes precede peripapillary retinal nerve fiber layer swelling 2. The acute thickening of these layers is then followed by long-term thinning. It has been suggested that this process is due to a state of pseudohypoxia, followed by a compensatory response 2. Optical coherence tomography angiography findings in patients with LHON have shown vascular dilation and tortuosity, which may also be seen clinically 6.
LHON inheritance
LHON is a mitochondrial disease and maternal inheritance is most common and this is known as mitochondrial LHON (mLHON). Rare autosomal recessive variants have been identified in those who do not have a mitochondrial mutation this is known as autosomal recessive LHON (arLHON).
Both mitochondrial LHON (mLHON) and autosomal recessive LHON (arLHON) are associated with sex-dependent incomplete penetrance. Relatives of the affected individual may or may not have developed visual loss. Only in approximately 60% of families with mitochondrial LHON (mLHON) is there a history of visual loss affecting the maternal relatives. In one nationwide cohort, the penetrance was reported to be 17.5% for males and 5.4% for females; the report concluded that the penetrance may be variant-specific. For patients with autosomal recessive LHON (arLHON), transmission is the same as any other autosomal recessive condition. Autosomal recessive is a pattern of inheritance where a genetic trait or condition is passed down when a child receives two copies of a mutated gene, one from each parent. An “autosomal” gene is located on one of the 22 non-sex chromosomes, and “recessive” means two copies of the altered gene are needed for the condition to manifest. Parents who carry one copy of the mutated gene are called carriers and typically do not have the condition themselves, but they have a 25% chance of having an affected child with each pregnancy.
Gene-specific penetrance risks for autosomal recessive LHON (arLHON) are yet to be reported but are likely to be similar to mitochondrial LHON (mLHON). Incomplete penetrance can be a challenge when counselling people, because most individuals who have a genetic variant associated with LHON will remain asymptomatic.
Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are both possible; however, both require caution because inaccurate interpretation of a positive prenatal test can occur due to the difference of mutational load in the sample as compared to the rest of the fetus. The presence of a LHON-causing genetic mutation does not predict whether a person will become symptomatic through life, or what the age of disease onset, rate of progression or depth of visual loss will be if they do become symptomatic.
LHON risk factors
Researchers don’t know exactly why some people with genetic mutations in the mitochondrial DNA (mtDNA) develop symptoms of LHON and others don’t. It is unknown why males are preferentially affected (up to 80% of patients); theories include the presence of a “susceptibility locus” on the Y chromosome. Some evidence suggests that physiological stress and environmental toxins may contribute to triggering the onset of symptoms. The theory is that these factors may add to the overall stress on body systems affected by LHON. Over time, they add up until they finally trigger symptoms.
Potential risk factors for LHON include:
- Smoking.
- Alcohol use.
- Exposure to environmental toxins.
- Systemic illness.
- Psychological stress.
Affected individuals should avoid smoking and excessive alcohol consumption, which generate reactive oxygen species (ROS) producing or amplifying mitochondrial impairments. One study found that patients with late-onset LHON had significantly higher cumulative tobacco and alcohol consumption than unaffected LHON mutation carriers, suggesting that these factors may in fact worsen disease severity once a person develops LHON 56.
LHON signs and symptoms
Leber hereditary optic neuropathy (LHON) usually begins as a one sided (unilateral) progressive optic nerve damage (optic neuropathy), with sequential involvement of your other eye months or years later 13. Symptoms typically begin in young adulthood and include blurred or cloudy vision, making reading, driving, and recognizing faces difficult, with vision loss often affecting one eye before the other. If vision is lost, then it usually occurs before 40 years of age.
LHON has traditionally been characterized into three major phases 57, 58:
- Asymptomatic phase
- Acute/subacute phase
- Atrophic phase.
During the asymptomatic phase, patients enjoy relatively good visual acuity; however, ophthalmoscopic examination may reveal circumpapillary vessel tortuosity and capillary microangiopathy in some asymptomatic patients 58. Furthermore, OCT of the optic nerve may demonstrate an increased Retinal Nerve Fiber Layer (RNFL) thickness of the temporal quadrant in some patients, which corresponds with the location of the papillomacular bundle 59. This increased thickness is thought to represent axoplasmic flow stasis and edema as a result of retinal ganglion cells (RGCs) injury, and suggests that there is selective and early involvement of the papillomacular bundle in LHON even in patients that are asymptomatic. It is currently unknown whether this Retinal Nerve Fiber Layer (RNFL) swelling heralds future clinical conversion.
The acute/subacute phase of LHON is characterized by a loss of central vision, including blurred vision and reduced perception of color. Individuals usually lose vision in one eye first and then lose vision in the other eye after two to three months typically reaching a nadir before entering the atrophic phase 60, 61. The acute/subacute phase of LHON has a classic triad on fundoscopic examination characterized by circumpapillary telangiectatic microangiopathy, swelling around the disc (pseudoedema), and lack of leakage on fluorescein angiography 57, 61. Longitudinal OCT studies of the optic nerve typically demonstrate significantly increased Retinal Nerve Fiber Layer (RNFL) thickness in all of the quadrants except the temporal quadrant during the acute/subacute phase 62. The less significant increase in the temporal RNFL is thought to be due to the preceding atrophy of the papillomacular bundle that occurs in the asymptomatic phase. Fundoscopic examination at three months demonstrates temporal pallor and disappearance of the triad observed in the acute/subacute phase 63.
At six months after onset of the acute/subacute phase, the patient enters the atrophic phase. The atrophic phase is characterized by bilateral optic atrophy and progression of visual field defects 9. Fundoscopic examination reveals diffuse pallor of the disc with no signs of edema. OCT shows a significant reduction in RNFL thickness in all quadrants. Within one year, most patients experience stabilization of their vision, but are often left with severe bilateral vision deficits and are legally blind. Recovery of vision is unlikely at this point in the disease process.
The chronic phase begins after 12 months, by which point visual acuity (VA) loss remains severe and permanent in the majority of cases, resulting in lifelong blindness 9, 10, 64.
Depending on the mutation and pedigree, most female carriers do not lose vision but up to half of males do.
Individuals eventually diagnosed with LHON may initially be asymptomatic or experience mild blurring of the central visual field of one eye. Males are more likely to be affected than females, with some reported ratios as high as 9:1. The age of onset is typically between 10 and 30 years, though onset in the seventh or eighth decades has been noted. Symptoms may progress from mild unilateral visual loss to severe bilateral visual loss, usually worse than 20/200.
Visual field testing typically shows a worsening central or cecocentral scotoma. The examination may initially reveal normal or pseudo-edematous optic nerves, with subsequent optic nerve pallor and atrophy.
“Leber plus” or “LHON Plus” disease describes patients with the clinical features of LHON (Leber’s hereditary optic neuropathy) in combination with other serious systemic or neurological abnormalities. These abnormalities include: postural tremor, motor disorder, multiple sclerosis-like syndrome, spinal cord disease, nonspecific myopathy (a broad term for a muscle disorder that involves muscle weakness, fatigue, or stiffness that doesn’t fit into a specific category and isn’t caused by inflammation), Parkinsonism with dystonia, anarthria (a severe motor speech disorder characterized by the complete inability to speak due to a lack of muscle control for articulation, often resulting from brain injury or neurological disease), dystonia (a movement disorder that causes muscular spasm and abnormal posture), motor and sensory peripheral neuropathy, spasticity, heart conduction abnormalities and mild encephalopathy (brain dysfunction) 14.
LHON diagnosis
LHON must be distinguished from other causes of optic neuropathy (optic nerve damage), as the treatment course may vary significantly. Individuals with LHON typically present with slowly progressive, painless visual blurring in one eye in the second to fourth decade of life, though the peak age of onset for all cases is the second and third decades of life 65. Studies have found the average age of onset to be 22–24 years in patients with the G11778A mutation and about 20 years in patients with the T14484C mutation 66, 67, 68.
Clinical suspicion for LHON may be heightened in the setting of a known family history of mitochondrial disease. The appearance of characteristic optic nerve changes such as blurred vision, blind spots, and reduced color vision, may lead to a tentative LHON diagnosis, but this can be confirmed with genetic testing. Late-phase LHON may display optic atrophy or optic nerve head pallor, which may be indistinguishable from other causes of optic neuropathy.
Signs and symptoms of LHON are often confused with those of optic neuritis (inflammation of an optic nerve), except for the lack of pain. Some patients may discover visual loss by incidentally covering the other eye, or by noting increasing difficulty with day-to-day tasks; other patients may present with LHON symptoms after the involvement of the second eye has begun (weeks to months later) and may thus complain of visual loss in both eyes. The longest interval reported between first and second eye involvement is 18 years 2. Other patients may note color or light desaturation. History of concurrent, LHON Plus–related neurologic or heart dysfunction should be elicited, including dystonia, tremor, neuropathy, movement disorders, weakness, nonspecific myopathy, and arrhythmias 2. Leigh syndrome may also be associated with LHON 68.
A history of worsening headaches, trauma, nutritional deficits (alcohol abuse), previous demyelinating disease, cancer, infiltrative orbital or lung disease, sexually transmitted disease, recent travel, or disease exposure may identify other causes of optic neuropathy. A family history of visual loss in maternal family members may be present.
Physical Examination
Visual acuity may be mildly reduced in the early stages of LHON, but can later progress to the counting fingers stage in one or both eyes 2. An afferent pupillary defect may be noted when LHON disease activity is asymmetric, often in the interval between the presentation of visual loss in one eye and progression to the other eye. Color testing reveals decreased red-green discrimination 2. Extraocular movement is typically intact, and confrontation visual fields may show intact peripheral vision 2. However, central or cecocentral scotoma (a type of vision loss that occurs in the central part of the visual field) may be described and more plainly delineated with formal visual field testing (Humphrey or Goldmann) 2. In patients presenting with symptoms in one eye, formal visual field testing may display subclinical visual deficits in the other eye, seemingly unaffected eye. Reduced contrast sensitivity and subnormal electroretinograms may also be present.
Slit lamp examination of the anterior segment is usually normal. Dilated fundus examination may be normal, or reveal hyperemic, pseudo-edematous optic nerves with peripapillary telangiectasias. Tortuosity of retinal arterioles may also be present. Though the optic nerve heads appear elevated and swollen, they do not display leakage on fluorescein angiogram, as opposed to those affected by inflammatory disease.
Diagnostic Procedures
A fluorescein angiogram may be performed to rule out true optic nerve swelling (edema). No dye leakage is noted along the borders of an otherwise swollen-appearing optic nerve head. Optical coherence tomography of the optic nerve may show elevation in the initial phases of the disease or atrophy in the later phases.
Laboratory Testing and Imaging
Based on patient history and physical examination, other causes of optic neuropathy should be excluded, especially in those with no family history of LHON. This may include laboratory serum testing of vitamin B12, folate (vitamin B9), rapid plasma reagin (RPR), fluorescent treponemal antibody absorption (FTA-ABS), antinuclear antibody (ANA), Lyme titers, angiotensin-converting enzyme, and/or purified protein derivative (PPD).
Magnetic resonance imaging (MRI) is especially recommended to rule out demyelinating disease and compressive lesions. Patients with LHON may have increased T2 signal in the optic nerves, chiasm, and tracts, so the orbital fat-suppressed contrast-enhanced MRI may show optic nerve enhancement similar to optic neuritis, as well as chiasmal enlargement and enhancement. Optic nerve enhancement and enlargement of the optic chiasm may be detected on MRI in LHON. In appropriate clinical scenarios, MRI findings should not dissuade physicians from including LHON in the differential diagnosis of acute optic neuropathy.
If other causes of optic neuropathy are comfortably excluded, gene testing can be performed via targeted mutation analysis. Ninety percent of patients with LHON patients harbor one of the three most common mutations, G11778A (guanine to adenine at position 11778), T14484C (tyrosine to cytosine at position 14484), and G3460A (guanine to adenine at position 3460) mutation. Identification of the mutations may help predict disease prognosis.
LHON differential diagnosis
Symptoms of the following disorders are similar to LHON. Comparisons may be useful for a differential diagnosis.
Autosomal dominant optic atrophy (DOA) is related to the OPA1 family of genes that influences mitochondrial biogenesis and function. Autosomal dominant optic atrophy (DOA) is an autosomal dominant genetic condition which means only one copy of a mutated gene from one parent is enough to cause the condition. “Autosomal” means the gene is on one of the non-sex chromosomes (numbered 1-22), and “dominant” means only one copy of the altered gene is necessary for the trait or disorder to be expressed. There is a 50% chance for a child to inherit the condition from an affected parent, and men and women are equally likely to be affected. The symptoms tend to come on at an earlier age (6-9) than in LHON and to do so in a more insidious and milder way. Generally, before puberty the child develops slowly progressive loss of vision and central scotomas a mild optic atrophy that progresses until about 20 years of age to a moderate loss of vision and optic atrophy. This difference in tempo and symmetry as well as the different genetics allows for the distinction with LHON.
Toxic or nutritional deficiencies can also produce bilateral mitochondrial optic neuropathy. There are several problems with metabolism that can lead to mitochondrial dysfunction that produces an acquired optic neuropathy that clinically seems very similar to these two genetic conditions (LHON, DOA).
LHON treatment
There is no cure for LHON. Management of LHON is primarily supportive, with an early introduction to visual aids and occupational therapy. Low-vision aids should be provided early. There is no established medical treatment for LHON, though there is a theoretical benefit from using antioxidants to help reduce the neurotoxic stress due to reactive oxygen species. Clinical trials are looking at the use of antioxidant supplements and gene therapy. Coenzyme Q10, L-carnitine, creatine, lipoic acid, lutein, dimethylglycine, cysteine, succinate, dichloroacetate, vitamin K1, vitamin K3, vitamin C, vitamin B1, vitamin B2, and vitamin E have all been suggested as treatment for LHON, but there is currently insufficient evidence to support their use 28, 29, 30. Several studies have shown that therapies involving ubiquinone and idebenone may provide possible benefits during both the acute and chronic phases of the disorder 31, 32.
Idebenone, a short-chain synthetic benzoquinone related to Coenzyme-Q10, is thought to restore mitochondrial function by bypassing the dysfunctional Complex I and electron transport directly to Complex III thus restoring mitochondrial adenosine triphosphate (ATP) generation and by acting as a potent antioxidant 33, 34, 35, 36. Recently, additional modes of action have been proposed, including effects on apoptosis, mitophagy, and myelination 37, 38.
Idebenone has undergone clinical testing for the treatment of several neurologic diseases. The recently completed RHODOS (Rescue of Hereditary Optic Disease Outpatient Study) was a double-blind, randomized, placebo-controlled trial of idebenone for patients with LHON and disease onset ≤5 years 39, 21. Eighty-five patients with one of the three primary LHON mutations and onset of symptoms within the last 5 years were randomized to receive idebenone 300 mg 3 times daily (900 mg/day) or placebo for 24 weeks (6 months) 39, 21. Though the study did not reach its primary endpoint (best recovery in visual acuity), a post hoc analysis revealed improvement in visual acuity in patients with discordant vision between eyes. Those with the G11778A and G3460A mutations seemed to derive greater benefit from idebenone. The drug was also found to be well tolerated. A trend toward improved visual acuity (VA) was observed in idebenone-treated patients. In hindsight, the 6-month treatment duration was likely too short to fully capture the potential treatment benefit. An expanded access program allowed for analysis of long-term idebenone treatment in the real world, in subacute/dynamic LHON patients (≤1 year after onset) 40. This noncontrolled study indicated the potential benefit of maintaining idebenone therapy for 24 to 30 months before classifying patients as nonresponders. This approach resulted in a visual acuity (VA) stabilization and/or recovery rate that was higher than expected from limited natural history studies 41, 42. Based on this cumulative clinical evidence, the European Medicines Agency approved idebenone (Raxone) for the treatment of individuals ≥12 years old with LHON 43. Raxone (idebenone) can only be obtained with a prescription and treatment should be started and supervised by a doctor with experience in LHON. Raxone (idebenone) is available as 150 mg tablets, and the recommended dose is two tablets taken three times a day with food 43. Long-term efficacy studies for idebenone in LHON are limited by the lack of direct control data, which are difficult to prospectively compile for rare diseases with an approved treatment. In addition, little data have been collected in the chronic phase (>1 year after onset) 44, 45.
An open-label, international, multicenter, natural history-controlled LEROS study assesses the efficacy and safety of idebenone treatment (900 mg/day) in patients with LHON up to 5 years after symptom onset (N = 199) and over a treatment period of 24 months, compared to an external natural history control cohort (N = 372), matched by time since symptom onset 46, 47. The LEROS study meets its primary endpoint based on clinically relevant benefit at month 12 and confirms the long-term efficacy of idebenone in the subacute/dynamic and chronic phases; the treatment effect varies depending on disease phase and the causative mtDNA mutation. LEROS confirmed the benefit of idebenone in LHON, including in the chronic phase (1–5 years since onset) with a consistent treatment benefit observed for patients with the most common G11778A (guanine to adenine at position 11778) mutation regardless of disease phase, and for patients with the T14484C (tyrosine to cytosine at position 14484) mutation in the chronic phase. LEROS confirms a favorable safety profile of idebenone in LHON patients. Howver, further study of idebenone use in patients carrying the G3460A (guanine to adenine at position 3460) mutation is needed to clarify treatment benefits. In the meantime, patients carrying G3460A mtDNA mutation who are in the subacute/dynamic phase should be adequately counseled to allow them to make an informed decision as to whether treatment with idebenone should be initiated. The findings of the LEROS study will help guide the clinical management of patients with LHON 47.
A second drug, the antioxidant alpha-tocotrienol-quinone (EPI-743), a vitamin E derivative, has also shown promising early results and may have more potency than idebenone. A small open-label study of five patients with acute LHON showed that early treatment with EPI-743 arrested disease progression in four out of five the LHON-patients 48, 49. However, a double-blind, randomized placebo-controlled trial is needed to provide confirmation of EPI-743 in the treatment of acute LHON.
Currently the most exciting potential means of treating LHON are within the field of gene therapy. There are several forms of gene therapy which are being evaluated as possible treatments for LHON, at various stages of testing.
The introduction of exogenous DNA into the mitochondrial genome with mitochondrial targeting of viral vectors is also promising. Recently, researchers conducted a phase 1 trial using intravitreal injection to deliver genetically modified adeno-associated viral vectors with the ND4 gene to affected eyes in 15 patients with LHON with the G11778A mutation. This therapy appeared to be safe and well-tolerated, with mostly mild intraocular inflammation. Another study of 5 patients with the G11778A mutation demonstrated similar results with respect to safety as well as some improvement in certain patients. However, it should be noted that these are the preliminary results of ongoing trials 6, 50, 51.
Idebenone
It is recommended that idebenone be started as soon as possible at 900 mg/day in patients with LHON still in the subacute/dynamic phase 7. Treatment should be continued for at least 1 year to assess the therapeutic response 7. A clinically relevant response (recovery of vision) to treatment should be defined according to an improvement of 2 lines of best-corrected visual acuity on Early Treatment Diabetic Retinopathy Study (ETDRS) charts or from off-chart to on-chart and an automated visual field test (mean deviation) 7. Once a favorable clinically relevant outcome has been confirmed, the treatment should be continued for another year 7.
Can glasses help?
Glasses help the eye focus light properly, usually when someone is short-sighted or long-sighted. They fix problems with how the eye bends light, not how the eye connects to the brain.
But LHON is different. LHON affects the optic nerve, which sends messages from the eyes to the brain. In LHON, the eyes can look healthy, but the messages don’t get through properly. Because of this, glasses usually don’t help improve or stop vision loss.
Medical Follow-Up
Individuals with LHON may be followed with serial visual acuity, visual field, contrast sensitivity, and color testing. The ideal recommended frequency of follow-up is approximately every 3 months for subacute and dynamic cases, then approximately every 6 months during the second year after the disease onset, and once a year after that.
LHON prognosis
It is difficult to predict who may lose vision. There is strong evidence that environmental factors such as smoking and excessive alcohol consumption are linked to why some people with LHON develop vision loss, and others do not 3. Therefore, a healthy lifestyle should be adopted by those at risk. This includes:
- not smoking
- drinking alcohol in moderation
- eating lots of fresh fruits and vegetables
- wearing head protection in contact sports.
In most patients with LHON, vision loss is likely to be rapid, severe and permanent 7. Most patients with LHON will develop optic atrophy at the end of one year. These patients suffer from marked visual impairment in their productive years of life that is irreversible. Most people with LHON will have to learn a new way of living with low vision. Low vision means moderate to severe visual impairment, but not total blindness. Moderate low vision scores 20/70 on a visual acuity test, while severe low vision scores 20/200 or worse. This is still a fairly wide range of visual acuity that you could end up with. However, some spontaneous recovery may occur gradually over 6 months to 1 year after an initial visual loss or may suddenly occur up to 10 years after onset 7. It may take the form of a gradual clearing of central vision or be restricted to a few central degrees, resulting in a small island of vision within a large central scotoma, which can be demonstrated on visual field testing 69.
Whether you end up on the moderate or severe side of this range is mostly a matter of luck. A good visual outcome is strongly correlated with a young age of onset 7. Most patients whose onset is before 20 years have a final visual acuity better than 20/80 7. Furthermore, the particular mtDNA mutation also influences prognosis, with the G11778A mutations carrying the worst overall prognosis for vision (only 4% reported spontaneous recovery) and the T14484C mutations the best (37% to 65% reported spontaneous recovery if the visual loss occurs before the age of 20) 7. The ultimate visual acuities in patients with the T14484C mutations are significantly better than those with the G11778A and G3460A mutations 7. Recurrences of vision loss are rare among patients, both with and without visual recovery 7. Treatment might make some difference, but the gene mutation you have makes the most difference. Some people with LHON recover some of their vision unexpectedly after a year of decline. Different gene mutations have different chances of recovery. But even with some recovery, you’ll likely still have low vision.
- Leber Hereditary Optic Neuropathy. https://rarediseases.org/rare-diseases/leber-hereditary-optic-neuropathy[↩][↩]
- Leber Hereditary Optic Neuropathy. https://eyewiki.org/Leber_Hereditary_Optic_Neuropathy[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Mackey DA, Ong JS, MacGregor S, Whiteman DC, et al. Is the disease risk and penetrance in Leber hereditary optic neuropathy actually low? Am J Hum Genet. 2023 Jan 5;110(1):170-176. doi: 10.1016/j.ajhg.2022.11.014[↩][↩]
- Manickam AH, Michael MJ, Ramasamy S. Mitochondrial genetics and therapeutic overview of Leber’s hereditary optic neuropathy. Indian J Ophthalmol. 2017 Nov;65(11):1087-1092. doi: 10.4103/ijo.IJO_358_17[↩]
- Gueven N, Nadikudi M, Daniel A, Chhetri J. Targeting mitochondrial function to treat optic neuropathy. Mitochondrion. 2017 Sep;36:7-14. doi: 10.1016/j.mito.2016.07.013[↩]
- Peragallo JH, Newman NJ. Is there treatment for Leber hereditary optic neuropathy? Curr Opin Ophthalmol. 2015 Nov;26(6):450-7. doi: 10.1097/ICU.0000000000000212[↩][↩][↩][↩]
- Shemesh A, Sood G, Blair K, et al. Leber Hereditary Optic Neuropathy (LHON) [Updated 2024 Mar 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482499[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Leber hereditary optic neuropathy. https://medlineplus.gov/genetics/condition/leber-hereditary-optic-neuropathy[↩]
- Carelli V., Carbonelli M., de Coo I.F., Kawasaki A., Klopstock T., Lagrèze W.A., La Morgia C., Newman N.J., Orssaud C., Pott J.W.R., et al. International consensus statement on the clinical and therapeutic management of Leber hereditary optic neuropathy. J. Neuro Ophthalmol. 2017;37:371–381. doi: 10.1097/WNO.0000000000000570[↩][↩][↩]
- Zeviani M., Carelli V. Mitochondrial retinopathies. Int. J. Mol. Sci. 2021;23 doi: 10.3390/ijms23010210[↩][↩][↩][↩]
- Yu-Wai-Man P., Griffiths P.G., Hudson G., Chinnery P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2009;46:145–158. doi: 10.1136/jmg.2007.054270[↩][↩][↩][↩]
- Smith AM, Czyz CN. Neuroanatomy, Cranial Nerve 2 (Optic) [Updated 2022 Nov 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507907[↩][↩]
- Lam BL, Feuer WJ, Abukhalil F, Porciatti V, Hauswirth WW, Guy J. Leber hereditary optic neuropathy gene therapy clinical trial recruitment: year 1. Arch Ophthalmol. 2010 Sep;128(9):1129-35. doi: 10.1001/archophthalmol.2010.201[↩][↩]
- Leber plus disease. https://www.orpha.net/en/disease/detail/99718[↩][↩]
- Riordan-Eva P., Sanders M.D., Govan G.G., Sweeney M.G., Da Costa J., Harding A.E. The clinical features of Leber’s hereditary optic neuropathy defined by the presence of a pathogenic mitochondrial DNA mutation. Brain. 1995;118:319–337. doi: 10.1093/brain/118.2.319[↩][↩]
- Jiang P., Liang M., Zhang J., Gao Y., He Z., Yu H., Zhao F., Ji Y., Liu X., Zhang M., et al. Prevalence of Mitochondrial ND4 Mutations in 1281 Han Chinese Subjects With Leber’s Hereditary Optic Neuropathy. Invest. Ophthalmol. Vis. Sci. 2015;56:4778–4788. doi: 10.1167/iovs.14-16158[↩][↩]
- Yen M.-Y., Wang A.-G., Wei Y.-H. Leber’s hereditary optic neuropathy: a multifactorial disease. Prog. Retin. Eye Res. 2006;25:381–396. doi: 10.1016/j.preteyeres.2006.05.002[↩][↩]
- Chalmers R.M., Harding A.E. A case-control study of Leber’s hereditary optic neuropathy. Brain. 1996;119:1481–1486. doi: 10.1093/brain/119.5.1481[↩][↩]
- Jiang P., Liang M., Zhang C., Zhao X., He Q., Cui L., Liu X., Sun Y.-H., Fu Q., Ji Y., et al. Biochemical evidence for a mitochondrial genetic modifier in the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum. Mol. Genet. 2016;25:3613–3625. doi: 10.1093/hmg/ddw199[↩][↩]
- Cheng H.-C., Chi S.-C., Liang C.-Y., Yu J.-Y., Wang A.-G. Candidate Modifier Genes for the Penetrance of Leber’s Hereditary Optic Neuropathy. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms231911891[↩][↩]
- Klopstock T, Yu-Wai-Man P, Dimitriadis K, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011 Sep;134(Pt 9):2677-86. doi: 10.1093/brain/awr170[↩][↩][↩][↩][↩][↩][↩]
- Newman NJ. Treatment of Leber hereditary optic neuropathy. Brain. 2011 Sep;134(Pt 9):2447-50. doi: 10.1093/brain/awr192[↩][↩][↩][↩]
- Newman N.J., Yu-Wai-Man P., Biousse V., Carelli V. Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: towards improved diagnosis and management. Lancet Neurol. 2023;22:172–188. doi: 10.1016/S1474-4422(22)00174-0[↩][↩]
- Stenton S.L., Sheremet N.L., Catarino C.B., Andreeva N.A., Assouline Z., Barboni P., Barel O., Berutti R., Bychkov I., Caporali L., et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Invest. 2021;131 doi: 10.1172/JCI138267[↩][↩]
- Gerber S., Ding M.G., Gérard X., Zwicker K., Zanlonghi X., Rio M., Serre V., Hanein S., Munnich A., Rotig A., et al. Compound heterozygosity for severe and hypomorphic NDUFS2 mutations cause non-syndromic LHON-like optic neuropathy. J. Med. Genet. 2017;54:346–356. doi: 10.1136/jmedgenet-2016-104212[↩][↩]
- Kieninger S., Xiao T., Weisschuh N., Kohl S., Rüther K., Kroisel P.M., Brockmann T., Knappe S., Kellner U., Lagrèze W., et al. DNAJC30 disease-causing gene variants in a large Central European cohort of patients with suspected Leber’s hereditary optic neuropathy and optic atrophy. J. Med. Genet. 2022;59:1027–1034. doi: 10.1136/jmedgenet-2021-108235[↩][↩]
- Fiorini C., Degiorgi A., Cascavilla M.L., Tropeano C.V., La Morgia C., Battista M., Ormanbekova D., Palombo F., Carbonelli M., Bandello F., et al. Recessive MECR pathogenic variants cause an LHON-like optic neuropathy. J. Med. Genet. 2023;61:93–101. doi: 10.1136/jmg-2023-109340[↩][↩]
- Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55:299–334. doi: 10.1016/j.survophthal.2009.10.002[↩][↩]
- Yu-Wai-Man P, Vortuba M, Moore AT, Chinnery PF. Treatment strategies for inherited optic neuropathies: past, present and future. Eye. 2014;28:521–537. doi: 10.1038/eye.2014.37[↩][↩]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012;18(4):CD004426. doi: 10.1002/14651858.CD004426.pub3[↩][↩]
- Amore G., Romagnoli M., Carbonelli M., Barboni P., Carelli V., La Morgia C. Therapeutic Options in Hereditary Optic Neuropathies. Drugs. 2021;81:57–86. doi: 10.1007/s40265-020-01428-3[↩][↩]
- Chen B.S., Yu-Wai-Man P., Newman N.J. Developments in the Treatment of Leber Hereditary Optic Neuropathy. Curr. Neurol. Neurosci. Rep. 2022;22:881–892. doi: 10.1007/s11910-022-01246-y[↩][↩]
- Gueven N., Faldu D. Idebenone treatment in Leber’s hereditary optic neuropathy: rationale and efficacy. Expert Opin. Orphan Drugs. 2013;1:331–339. doi: 10.1517/21678707.2013.772894[↩][↩]
- Haefeli R.H., Erb M., Gemperli A.C., Robay D., Courdier Fruh I., Anklin C., Dallmann R., Gueven N. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS One. 2011;6 doi: 10.1371/journal.pone.0017963[↩][↩]
- Rauchova H, Vrbacky M, Bergamini C, et al. Inhibition of glycerophosphate-dependent H2O2 generation in brown fat mitochondria by idebenone. Biochem Biophys Res Commun. 2006;339:362–366. doi: 10.1016/j.bbrc.2005.11.035[↩][↩]
- Haefeli RH, Erb M, Gemperli AC, et al. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS One. 2011;6:e17963. doi: 10.1371/journal.pone.0017963[↩][↩]
- Gueven N., Ravishankar P., Eri R., Rybalka E. Idebenone: When an antioxidant is not an antioxidant. Redox Biol. 2021;38 doi: 10.1016/j.redox.2020.101812[↩][↩]
- Danese A., Patergnani S., Maresca A., Peron C., Raimondi A., Caporali L., Marchi S., La Morgia C., Del Dotto V., Zanna C., et al. Pathological mitophagy disrupts mitochondrial homeostasis in Leber’s hereditary optic neuropathy. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111124[↩][↩]
- Study to Assess Efficacy,Safety and Tolerability of Idebenone in the Treatment of Leber’s Hereditary Optic Neuropathy (RHODOS). https://clinicaltrials.gov/study/NCT00747487[↩][↩][↩]
- Catarino C.B., von Livonius B., Priglinger C., Banik R., Matloob S., Tamhankar M.A., Castillo L., Friedburg C., Halfpenny C.A., Lincoln J.A., et al. Real-world clinical experience with idebenone in the treatment of Leber hereditary optic neuropathy. J. Neuro Ophthalmol. 2020;40:558–565. doi: 10.1097/WNO.0000000000001023[↩][↩]
- Newman N.J., Carelli V., Taiel M., Yu-Wai-Man P. Visual outcomes in Leber hereditary optic neuropathy patients with the m.11778GA (MTND4) mitochondrial DNA mutation. J. Neuro Ophthalmol. 2020;40:547–557. doi: 10.1097/WNO.0000000000001045[↩][↩]
- Yu-Wai-Man P., Newman N.J., Carelli V., La Morgia C., Biousse V., Bandello F.M., Clermont C.V., Campillo L.C., Leruez S., Moster M.L., et al. Natural history of patients with Leber hereditary optic neuropathy-results from the REALITY study. Eye (Lond) 2022;36:818–826. doi: 10.1038/s41433-021-01535-9[↩][↩]
- Raxone. https://www.ema.europa.eu/en/medicines/human/EPAR/raxone[↩][↩][↩][↩]
- Pemp B., Kircher K., Reitner A. Visual function in chronic Leber’s hereditary optic neuropathy during idebenone treatment initiated 5 to 50 years after onset. Graefes Arch. Clin. Exp. Ophthalmol. 2019;257:2751–2757. doi: 10.1007/s00417-019-04444-6[↩][↩]
- Pemp B., Mitsch C., Kircher K., Reitner A. Changes in visual function and correlations with inner retinal structure in acute and chronic Leber’s hereditary optic neuropathy patients after treatment with idebenone. J. Clin. Med. 2021;10:151. doi: 10.3390/jcm10010151[↩][↩]
- Study to Assess the Efficacy and Safety of Raxone in LHON Patients (LEROS). https://clinicaltrials.gov/study/NCT02774005[↩][↩]
- Yu-Wai-Man P, Carelli V, Newman NJ, et al. LEROS Study Group. Therapeutic benefit of idebenone in patients with Leber hereditary optic neuropathy: The LEROS nonrandomized controlled trial. Cell Rep Med. 2024 Mar 19;5(3):101437. doi: 10.1016/j.xcrm.2024.101437[↩][↩][↩][↩]
- Sadun AA, Chicani CF, Ross-Cisneros FN, et al. Effect of EPI-743 on the Clinical Course of the Mitochondrial Disease Leber Hereditary Optic Neuropathy. Arch Neurol. 2012;69(3):331–338. doi:10.1001/archneurol.2011.2972[↩][↩]
- Emergency Administration of EPI-743 to a Single Patient With Leber’s Hereditary Optic Neuropathy [LHON]. https://clinicaltrials.gov/study/NCT02300753[↩][↩]
- Feuer WJ, Schiffman JC, Davis JL, Porciatti V, Gonzalez P, Koilkonda RD, Yuan H, Lalwani A, Lam BL, Guy J. Gene Therapy for Leber Hereditary Optic Neuropathy: Initial Results. Ophthalmology. 2016 Mar;123(3):558-70. doi: 10.1016/j.ophtha.2015.10.025[↩][↩]
- Bouquet C, Vignal Clermont C, Galy A, Fitoussi S, Blouin L, Munk MR, Valero S, Meunier S, Katz B, Sahel JA, Thomasson N. Immune Response and Intraocular Inflammation in Patients With Leber Hereditary Optic Neuropathy Treated With Intravitreal Injection of Recombinant Adeno-Associated Virus 2 Carrying the ND4 Gene: A Secondary Analysis of a Phase 1/2 Clinical Trial. JAMA Ophthalmol. 2019 Apr 1;137(4):399-406. doi: 10.1001/jamaophthalmol.2018.6902[↩][↩]
- Leber Hereditary Optic Neuropathy: A 17-year-old male presents with progressive, painless, bilateral vision loss. https://eyerounds.org/cases/172-LHON.htm#gsc.tab=0[↩]
- Hedges TR, Gobuty M, Manfready RA, Erlich-Malona N, Monaco C, Mendoza-Santiesteban CE. The Optical Coherence Tomographic Profile of Leber Hereditary Optic Neuropathy. Neuroophthalmology. 2016 May 2;40(3):107-112. doi: 10.3109/01658107.2016.1173709[↩]
- Marshall AS, Jones NS. Discovering Cellular Mitochondrial Heteroplasmy Heterogeneity with Single Cell RNA and ATAC Sequencing. Biology (Basel). 2021 Jun 5;10(6):503. doi: 10.3390/biology10060503[↩]
- Palace J. Multiple sclerosis associated with Leber’s Hereditary Optic Neuropathy. J Neurol Sci. 2009 Nov 15;286(1-2):24-7. doi: 10.1016/j.jns.2009.09.009[↩]
- van Westen D, Hammar B, Bynke G. Magnetic resonance findings in the pregeniculate visual pathways in Leber hereditary optic neuropathy. J Neuroophthalmol. 2011 Mar;31(1):48-51. doi: 10.1097/WNO.0b013e3181f3f203[↩]
- Nikoskelainen E, Sogg RL, Rosenthal AR, Friberg TR, Dorfman LJ. The Early Phase in Leber Hereditary Optic Atrophy. Arch Ophthalmol. 1977;95(6):969–978. doi:10.1001/archopht.1977.04450060055002[↩][↩]
- Nikoskelainen E, Hoyt WF, Nummelin K. Ophthalmoscopic Findings in Leber’s Hereditary Optic Neuropathy: II. The Fundus Findings in the Affected Family Members. Arch Ophthalmol. 1983;101(7):1059–1068. doi:10.1001/archopht.1983.01040020061011[↩][↩]
- Savini G, Barboni P, Valentino ML, Montagna P, Cortelli P, De Negri AM, Sadun F, Bianchi S, Longanesi L, Zanini M, Carelli V. Retinal nerve fiber layer evaluation by optical coherence tomography in unaffected carriers with Leber’s hereditary optic neuropathy mutations. Ophthalmology. 2005 Jan;112(1):127-31. doi: 10.1016/j.ophtha.2004.09.033[↩]
- Yen MY, Wang AG, Wei YH. Leber’s hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res. 2006 Jul;25(4):381-96. doi: 10.1016/j.preteyeres.2006.05.002[↩]
- Smith JL, Hoyt WF, Susac JO. Ocular Fundus in Acute Leber Optic Neuropathy. Arch Ophthalmol. 1973;90(5):349–354. doi:10.1001/archopht.1973.01000050351002[↩][↩]
- Barboni P, Savini G, Valentino ML, Montagna P, Cortelli P, De Negri AM, Sadun F, Bianchi S, Longanesi L, Zanini M, de Vivo A, Carelli V. Retinal nerve fiber layer evaluation by optical coherence tomography in Leber’s hereditary optic neuropathy. Ophthalmology. 2005 Jan;112(1):120-6. doi: 10.1016/j.ophtha.2004.06.034[↩]
- Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004 Jan;23(1):53-89. doi: 10.1016/j.preteyeres.2003.10.003[↩]
- Gueven N. Optic neurodegeneration: Time to act. Biol. Med. 2014;01 doi: 10.4172/0974-8369.S1-001[↩]
- Vaphiades MS. Magnetic resonance findings in the pregeniculate visual pathways in Leber hereditary optic neuropathy. J Neuroophthalmol. 2011 Jun;31(2):194; author reply 194. doi: 10.1097/WNO.0b013e31821bca86[↩]
- Phillips PH, Vaphiades M, Glasier CM, Gray LG, Lee AG. Chiasmal enlargement and optic nerve enhancement on magnetic resonance imaging in leber hereditary optic neuropathy. Arch Ophthalmol. 2003 Apr;121(4):577-9. doi: 10.1001/archopht.121.4.577[↩]
- Vaphiades MS, Phillips PH, Turbin RE. Optic nerve and chiasmal enhancement in leber hereditary optic neuropathy. J Neuroophthalmol. 2003 Mar;23(1):104-5. doi: 10.1097/00041327-200303000-00057[↩]
- Vaphiades MS, Newman NJ. Optic nerve enhancement in leber hereditary optic neuropathy: four years later. J Neuroophthalmol. 2002 Mar;22(1):66-7. doi: 10.1097/00041327-200203000-00037[↩][↩]
- Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies – disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011 Mar;30(2):81-114. doi: 10.1016/j.preteyeres.2010.11.002[↩]




{kind=link}