Contents
What is lissencephaly
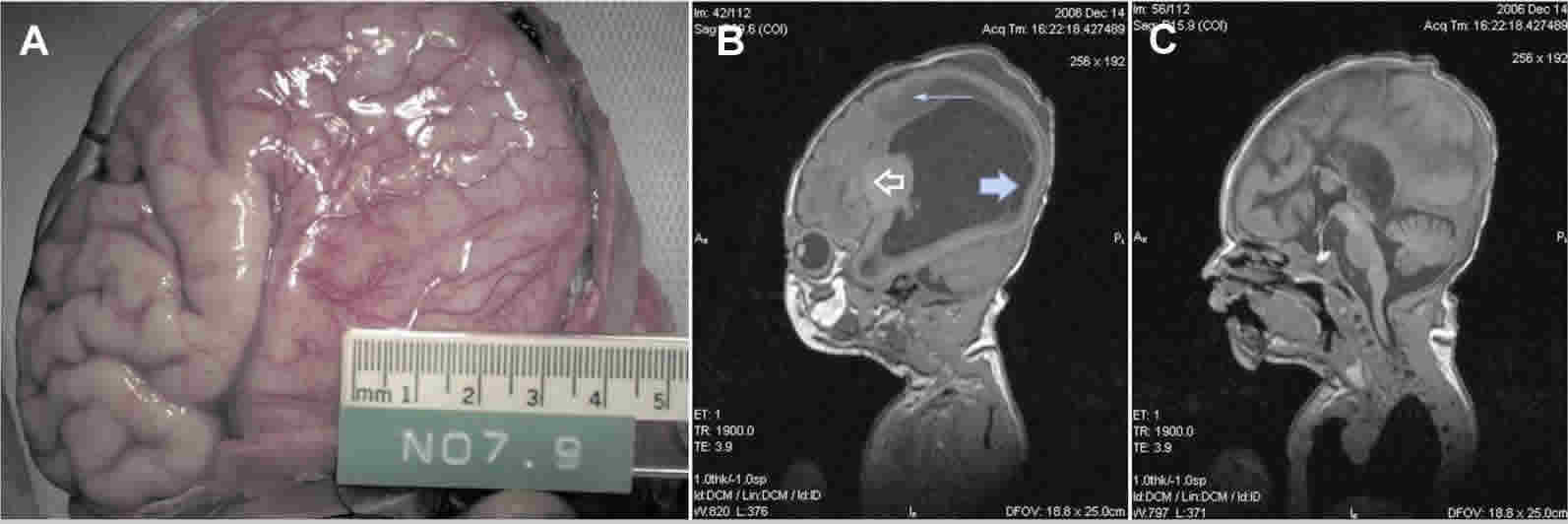
Lissencephaly, which literally means “smooth brain,” is a group of rare, gene-linked brain malformations characterized by the absence of normal brain convolutions (folds) in the cerebral cortex and an abnormally small head (microcephaly) 1). Lissencephaly is caused by defective neuronal migration during embryonic development, the process in which nerve cells move from their place of origin to their permanent location within the cerebral cortex gray matter. Microscopically, there is often disruption of the normal six-layer structure of the neocortex 2).
In the usual condition of lissencephaly, children usually have a normal sized head at birth. In children with reduced head size at birth, the condition microlissencephaly is typically diagnosed. Children with lissencephaly may have unusual facial appearance, feeding and swallowing problems, failure to thrive, muscle tone anomalies (early hypotonia and subsequently limb hypertonia), seizures (in particular, infantile spasms), and severe psychomotor retardation. Hands, fingers, or toes may be deformed. Lissencephaly may be associated with other diseases including isolated lissencephaly sequence, Miller-Dieker syndrome, and Walker-Warburg syndrome. Sometimes it can be difficult to distinguish between these conditions clinically so consultation with experts in lissencephaly is recommended to help ensure correct diagnosis and possible molecular testing.
Multiple forms of lissencephaly have been described and their current classification is based on the associated malformations and underlying cause.
Two large lissencephaly groups can be distinguished:
- Classic lissencephaly (also known as type 1 lissencephaly and its variants) and
- Cobblestone lissencephaly (also known as type 2 lissencephaly).
The incidence of all forms of type 1 lissencephaly (classical lissencephaly and its variants) is around 1 in 100,000 births 3).
Management of lissencephaly is symptomatic only (swallowing problems require adapted feeding to prevent food aspiration, articular and respiratory physiotherapy to prevent orthopedic problems resulting from hyptonia, and treatment of gastroesophageal reflux). The epilepsy is often resistant to treatment.
The following online resources can also help you find a genetics professional in your community:
- The National Society of Genetic Counselors (https://www.nsgc.org/) provides a searchable directory of US and international genetic counseling services.
- The American College of Medical Genetics (https://www.acmg.net/) has a searchable database of US genetics clinics.
- The University of Kansas Medical Center (http://www.kumc.edu/gec/prof/genecntr.html) provides a list of US and international genetic centers, clinics, and departments.
- The American Society of Human Genetics (http://www.ashg.org/membership/member_search.shtml) maintains a database of its members, which includes individuals who live outside the United States. Visit the link to obtain a list of the geneticists in your country, some of whom may be researchers that do not provide medical care.
Lissencephaly type 1
In lissencephaly type 1 also known as classic lissencephaly, the cortex appears thickened, with four more or less disorganized layers rather than six normal layers. Lissencephaly type 1 is a brain malformation that may occur as an isolated abnormality (isolated lissencephaly sequence) or in association with certain syndromes (e.g., Miller-Dieker syndrome). Lissencephaly type 1 is characterized by agyria or pachygyria, which means absence or incomplete development, respectively, of the brain gyri or convolution, causing the brain’s surface to appear unusually smooth. In the variants of lissencephaly type 1 (classical lissencephaly), extra-cortical anomalies are also present (total or subtotal agenesis of the corpus callosum and/or cerebellar hypoplasia).
Lissencephaly type 1 and the variant forms can be further divided into several subgroups. Four forms can be distinguished on the basis of their genetic cause:
- Anomalies in the LIS1 gene (isolated lissencephaly and Miller-Dieker syndrome),
- Anomalies in the TUBA3 and DCX genes, and
- Lissencephalies caused by mutations in the ARX gene (X-linked lissencephaly with agenesis of the corpus callosum (XLAG) syndrome).
In addition to these four entities, isolated lissencephalies without a known genetic defect, lissencephalies with severe microcephaly (microlissencephaly) and lissencephalies associated with polymalformative syndromes are also included in the group of classical lissencephalies.
Cobblestone lissencephaly (formally referred to as lissencephaly type type II) is present in three entities:
- the Walker-Warburg, Fukuyama and MEB (Muscle-Eye-Brain) syndromes.
- It is characterized by global disorganization of cerebral organogenesis with an uneven cortical surface (with a pebbled or cobblestone appearance).
- Microscopic examination reveals total disorganization of the cortex and the absence of any distinguishable layers.
Infants with classical lissencephaly may have a head that is smaller than would be expected (microcephalic). Additional abnormalities may include seizures, profound intellectual disability, feeding difficulties, growth retardation, and impaired motor abilities. If an underlying syndrome is present, there may be additional symptoms and physical findings.
Brain imaging
Brain magnetic resonance imaging (MRI) studies of all patients included T1- and T2-weighted sequences, and variable high-resolution (less than 2 mm) volumetric and other sequences obtained in sagittal, axial and usually coronal planes. All were reviewed with particular attention to the severity (lissencephaly grade) and gradient of the gyral malformation, cortical thickness and presence of associated brain malformations.
Agyria was defined as cortical regions with sulci >3 cm apart, pachygyria as abnormally wide gyri with sulci 1.5–3 cm apart, and subcortical band heterotopia as longitudinal bands of gray matter located deep to the cerebral cortex and separated from it by a thin layer of normal appearing white matter. The gyral pattern is characterized by abnormally shallow sulci. The cerebral cortex in agyria and pachygyria was either very thick, measuring 10–20 mm (thick or classic lissencephaly), or less often mildly thick measuring 5–10 mm (“thin” lissencephaly, see results). The term “dysgyria” for a cortical appearance intermediate between lissencephaly (pachygyria) and polymicrogyria consisting of mixed large and small gyri separated by shallow sulci with a smooth gray-white border.
Table 1. Imaging criteria used for lissencephaly-subcortical band heterotopia classification
|
|
|
|
Figure 1. Lissencephaly with subcortical band heterotopia
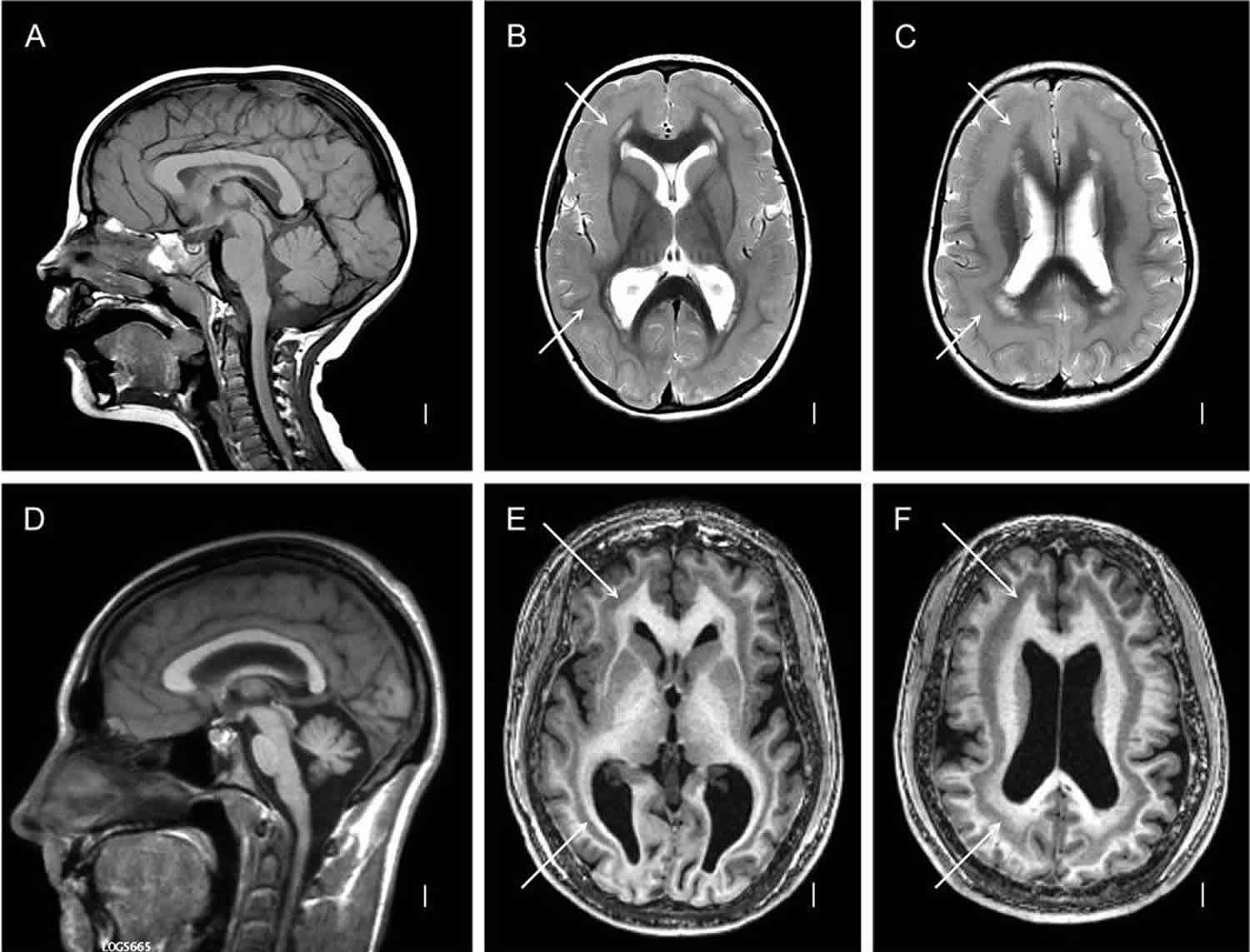
Footnote: (A–C) diffuse thick, (D–F) diffuse thin subcortical band heterotopia. T1-weighted midline sagittal (A, D) images are normal, except for mild cerebellar vermis hypoplasia in the second row (D). Axial T2-weighted (B–C) or three-dimensional (E–F) images through low or high lateral ventricles show the bands (white arrows) as well as simplified gyri with shallow sulci and a thin layer of white matter between the cortex and bands.
[Source 5) ]Lissencephaly prognosis
The prognosis for children with lissencephaly depends on the degree of brain malformation. The encephalopathy associated with lissencephaly is often very severe and affected children are completely dependent on their carer.
Many children will die before the age of 10 years. The cause of death is usually aspiration of food or fluids, respiratory disease, or severe seizures. Some will survive, but show no significant development — usually not beyond a 3- to 5-month-old level. Others may have near-normal development and intelligence. Because of this range, it is important to seek the opinion of specialists in lissencephaly and support from family groups with connection to these specialists.
Lissencephaly life expectancy
Many children will die before the age of 10 years. The cause of death is usually aspiration of food or fluids, respiratory disease, or severe seizures. Some will survive, but show no significant development — usually not beyond a 3- to 5-month-old level. Others may have near-normal development and intelligence. Because of this range, it is important to seek the opinion of specialists in lissencephaly and support from family groups with connection to these specialists.
The lissencephaly-subcortical band heterotopia severity grade generally correlates with clinical outcome, with severe lissencephaly (agyria) more severe than intermediate lissencephaly (pachygyria), and both more severe than subcortical band heterotopia. But when other imaging criteria are added, correlation with the clinical severity becomes less consistent, making clinical management more difficult. To address this need, we have correlated the major imaging patterns with the typical developmental outcome based on published reports and our extensive clinical experience, and adapted this to fit with the new classification scheme (Table 2). Each of three clinical severity grades – mild (1), moderate (2) and severe (3) – includes three to four morphological subtypes that have a different impact on the neurological status and mental development. Exceptions may occur with these subtypes, most frequently with severe epilepsy, which often lowers overall function.
To supplement the expanded imaging classification in Table 2, the major medical problems influencing the developmental prognosis and lowering the life expectancy are feeding difficulties that include gastro-esophageal reflux and aspiration, epilepsy of many different types, and pneumonia 6). 35–85% of children with classic (thick) lissencephaly develop infantile spams 7). Seizures are often intractable and many patients have the classic electrophysiological signs of Lennox-Gastaut syndrome. Cognitive development often slows with the onset of seizures 8).
Table 2. Clinical severity of lissencephaly
| Grade | Subtypes1 | Imaging pattern | Clinical outcome |
|---|---|---|---|
| 1. Mild | 1-1 | Partial subcortical band heterotopia a>p or p>a | Borderline to moderate intellectual disability, seizures of variable severity; survival into adulthood expected 9) |
| 1-2 | Diffuse thin subcortical band heterotopia (<10mm) | ||
| 1-3 | Partial pachygyria a>p or p>a | ||
| 1-4 | Isolated “thin” or undulating lissencephaly | ||
| 2. Moderate | 2-1 | Diffuse thick subcortical band heterotopia (>10mm) | Moderate to severe intellectual disability, severe language impairment, seizures often poorly controlled, life expectancy may be reduced although many survive to adulthood 10) |
| 2-2 | Mixed pachygyria-subcortical band heterotopia | ||
| 2-3 | Diffuse pachygyria a>p or p>a | ||
| 3. Severe | 3-1 | Mixed pachygyria-agyria | Profound intellectual disability, poorly controlled seizures, short survival typical with mortality rate ~50% by 10 years with normal cerebellum, and much higher with cerebellar hypoplasia 11) |
| 3-2 | Diffuse agyria | ||
| 3-3 | Agyria with cerebellar hypoplasia |
1Primary groups based on severity of intellectual disability and epilepsy, subtypes based on imaging characteristics.
[Source 12) ]Lissencephaly causes
Lissencephaly may be due to various non-genetic and genetic factors. Such factors may include intrauterine viral infections, insufficient supply of oxygenated blood to the brain (ischemia) during fetal development, and/or certain genetic mutations. Changes (mutations) in several genes have been implicated in isolated lissencephaly, to date 20 genes have been associated with lissencephaly 13): LIS1, RELN, TUBA1A, NDE1, KATNB1, CDK5, ARX and DCX. Of these, LIS1 and DCX 14) and TUBA1A 15) gene mutations have been most studied.
One of the best-studied examples is LIS1 or PAFAH1B1. Mutations in this gene are responsible for lissencephaly type 1. LIS1 gene is localized on chromosome 17p13.3. The gene encodes for platelet-activating factor acetylhydrolase isoform 1B that interacts with microtubule associated proteins: dynein and dynactin. This interaction is critical for proper neuronal migration during fetal brain development; disruption of this interaction results in lissencephaly. Most infants with isolated lissencephaly sequence show mutations or deletions of just the LIS1 gene, whereas infants with Miller-Dieker syndrome are mostly found to have mutations in the LIS1 gene but also have additional deletions of adjacent genes on chromosome 17, thus resulting in lissencephaly type 1 features and other craniofacial abnormalities. Such chromosomal alterations occur randomly and are observed in the child only, without evidence of alteration in either parent. Importantly, this genetic form of lissencephaly does not recur in families, and so the risk of another child with this condition is extremely low.
Of the genes that have been implicated in lissencephaly, DCX and ARX genes are notable because they are localized on the X chromosome. This genetic form of lissencephaly can be observed in more than one child per family, because the mutation can be present in the DNA of a healthy mother. Lissencephaly caused by DCX and ARX is referred to as X-linked lissencephaly type 1 and 2, respectively (XLIS 1-2 or LISX 1-2). Because males only have one X chromosome, males who inherit the disease gene are more likely to manifest the full spectrum of abnormalities associated with the disorder and therefore are usually more severely affected. Females who inherit this gene mutation may have a more variable presentation and be more mildly affected than the males, or can be healthy without symptoms.
The DCX gene encodes for the doublecortin protein. Doublecortin associates with microtubules to regulate neuronal migration. X-linked mutations may appear randomly or can be inherited. The ARX gene encodes for the aristaless-related homeobox protein. In addition to classic lissencephaly features, infants with a ARX mutation may also have absence of portions of the brain (hydranencephaly), abnormal genitalia, severe epilepsy and other abnormalities.
Other gene mutations that have been associated with lissencephaly, such as RELN, which causes Norman-Roberts syndrome, have an autosomal recessive inheritance pattern. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
In addition to LIS1, RELN, DCX and ARX, mutations in other genes have also been found to cause lissencephaly. They include: TUBA1A, NDE1, KATNB1, and CDK5. These genes share molecular function with LIS1 and DCX, working as part of the cellular machinery of dynein and dynactin required for neuronal migration during fetal brain development.
Emerging evidence suggests that genetic alterations and non-genetic causes result in lissencephaly due to impaired neuronal migration of the outer region of the brain during fetal development. The cerebral cortex, which is responsible for conscious movement and thought, normally consists of several deep gyri and sulci (grooves), which are formed by “in-folding” of the cerebral cortex. During embryonic growth, newly formed cells that will later develop into specialized nerve cells normally migrate to the brain’s surface (neuronal migration), resulting in the formation of several cellular layers. However, in cases of lissencephaly type 1, the cells fail to migrate to their destined locations resulting in neuronal dysmigration, and the cerebral cortex develops an insufficient number of cellular layers, with absence or incomplete development of gyri.
Lissencephaly symptoms
All patients with lissencephaly have intellectual disability, but the severity differs significantly based on the subtype of lissencephaly from profound disability and limited survival with diffuse agyria to mild intellectual or learning disability in patients with partial subcortical band heterotopia 16). Seizures occur in most patients with lissencephaly 17).
Newborns with lissencephaly type 1 who have no underlying syndrome are said to have isolated lissencephaly sequence. In addition to lissencephaly, those with the condition may have other associated brain malformations, such as absence or underdevelopment of the corpus callosum, which is the thick band of nerve fibers that join and carry messages between the brain’s two cerebral hemispheres. Affected infants often also have microcephaly, seizures, and severe or profound intellectual disability. In addition, those with the condition may have a normal facial appearance or subtle facial changes, such as a relatively small jaw (micrognathia) or a slight indentation of the temples (bitemporal hollowing). Additional symptoms and findings may include feeding difficulties, growth failure, abnormally diminished muscle tone (hypotonia) early in life, and increased muscle tone (hypertonia) later during infancy, and impaired motor abilities.
Lissencephaly type 1 also occurs in association with genetic syndromes, including Miller-Dieker syndrome and Norman-Roberts syndrome. In addition to signs and symptoms of classical lissencephaly, infants with Miller-Dieker syndrome may also malformations including microcephaly with a broad, high forehead; bitemporal hollowing; a relatively wide face; micrognathia; a long, thin upper lip; a short nose with upturned nostrils; low-set, malformed ears; polydactyly; abnormal palmar creases; cataracts and/or malformations of the heart, kidneys and/or other organs.
Norman-Roberts syndrome is also characterized by lissencephaly type 1 features with certain craniofacial abnormalities, such as a low, sloping forehead; abnormal prominence of the back portion of the head; a broad, prominent nasal bridge; and widely set eyes (ocular hyperterlorism).
Lissencephaly diagnosis
When the suspicion is high for lissencephaly type 1 because of family history and/or prenatal ultrasound screening, it is possible that the condition may be confirmed by specialized testing during pregnancy, such as cell-free fetal DNA, amniocentesis or chorionic villus sampling (CVS).
Lissencephaly type 1 may be diagnosed thorough clinical evaluation, brain imaging studies, including computerized tomography (CT) scanning and/or magnetic resonance imaging (MRI) and genetic testing like chromosomal analysis and/or specific gene mutational analysis. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of the brain’s tissue structure. With MRI, a magnetic field and radio waves create cross-sectional images of the brain. Another test that can aid in the diagnosis is electroencephalogram (EEG). During an EEG, the brain’s electrical impulses are recorded. Brain malformations, including lissencephaly, are often associated with abnormal brain electrical impulses and/or seizures. An abnormal EEG pattern may prompt further brain imaging and lead to the diagnosis of lissencephaly. Lastly, DNA analysis may detect certain deletions/mutations in genes linked to lissencephaly. Commercially available gene testing for known genetic causes of lissencephaly is now available. The number of genes included in these tests continues to expand with additional research.
Lissencephaly treatment
There is no cure for lissencephaly, but children can show progress in their development over time. Supportive care may be needed to help with comfort, feeding, and nursing needs. Seizures may be particularly problematic but anticonvulsant medications can help. Progressive hydrocephalus (an excessive accumulation of cerebrospinal fluid in the brain) is very rare, seen only in the subtype of Walker-Warburg syndrome, but may require shunting. If feeding becomes difficult, a gastrostomy tube may be considered.
The treatment of lissencephaly type 1 is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, and other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Therapies for individuals with lissencephaly type 1 are symptomatic and supportive. Treatment may include measures to improve the intake of nutrients in infants with feeding difficulties; the administration of anticonvulsant drugs to help prevent, reduce, or control seizures; and/or other measures.
Genetic counseling is recommended for families of affected children.
References [ + ]

{kind=link}