Contents
What is pheochromocytoma
Pheochromocytomas (also known as adrenal chromaffin tumor) are rare tumors that start in the inner section of the adrenal gland (the chromaffin cells of the adrenal medulla). Pheochromocytomas are mostly benign (noncancerous), but have a 10 percent rate of being cancerous (malignant). Pheochromocytomas occur most frequently in young to middle-aged adults (between the ages of 30 to 60). Approximately 10 percent of these involve both adrenal glands. Pheochromocytomas often cause the adrenal gland to make too many stress hormones called epinephrines (adrenaline) and norepinephrines (noradrenaline). Adrenaline and noradrenaline are hormones that trigger your body’s fight-or-flight response to a perceived threat. The hormones prompt higher blood pressure, a faster heart rate and a boost in other body systems that enable you to react quickly. A pheochromocytoma results in the irregular and excessive release of these hormones. This can lead to high blood pressure and cause symptoms such as severe headaches, irritability, sweating, rapid heart rate, nausea, vomiting, weight loss, weakness, chest pain, and anxiety. Rarely, this kind of pheochromocytoma tumor occurs outside the adrenal gland, usually somewhere in the abdomen. These are called extra-adrenal pheochromocytomas or paragangliomas. The cause of most pheochromocytomas is unknown. In some cases, there is a genetic cause. This type of tumor can occur in certain familial genetic syndromes, including multiple endocrine neoplasia, type 2 (MEN2), neurofibromatosis type 1, Von Hippel-Lindau disease, hereditary paraganglioma-pheochromocytoma syndrome, Carney triad, and Carney-Stratakis dyad. There are also several genes that have been associated with pheochromocytoma when it does not occur as part of a syndrome 1.
Pheochromocytoma is a rare tumor with an estimated rate of two to eight per million people per year. An incidentally discovered adrenal mass by CT scan, MRI or ultrasound is called an incidentaloma. Four to five percent of incidentalomas will be diagnosed as a pheochromocytoma by laboratory tests. This is the reason why every patient with an incidentally discovered adrenal mass should be tested for pheochromocytoma. However, in patients simply evaluated for high blood pressure, only 0.2 to 0.6 percent of adults and one percent of children would have pheochromocytoma as a cause.
Most people with a pheochromocytoma are between the ages of 20 and 50, but the tumor can develop at any age. Treatment depends on several factors, including the size of your tumor, whether it has spread and your general health and fitness. Surgical treatment to remove a pheochromocytoma usually returns blood pressure to normal.
Pheochromocytoma triad
Carney triad 2 is an extremely rare disorder that primarily affects young women. As initially described in 1977 3, the classic Carney triad included extra-adrenal sympathetic paraganglioma, gastric stromal sarcoma, and pulmonary chondroma. Adrenal cortical adenoma and esophageal leiomyoma were later shown to be associated with the syndrome 4. Carney found that 78% of affected individuals had two of the three classic tumors and 22% had all three neoplasms 5. No pathogenic variants in genes associated with hereditary paragangliomas or hereditary gastrointestinal stromal tumors have been found in any of the reported individuals 4. However, chromosomal changes that appeared to correlate with the syndrome, including possible loss of regions on the short arm (1p) and the long arm (1q) of chromosome 1, were noted. The additional neoplasms associated with this syndrome should differentiate it from the hereditary paraganglioma-pheochromocytoma syndromes.
The adrenal glands
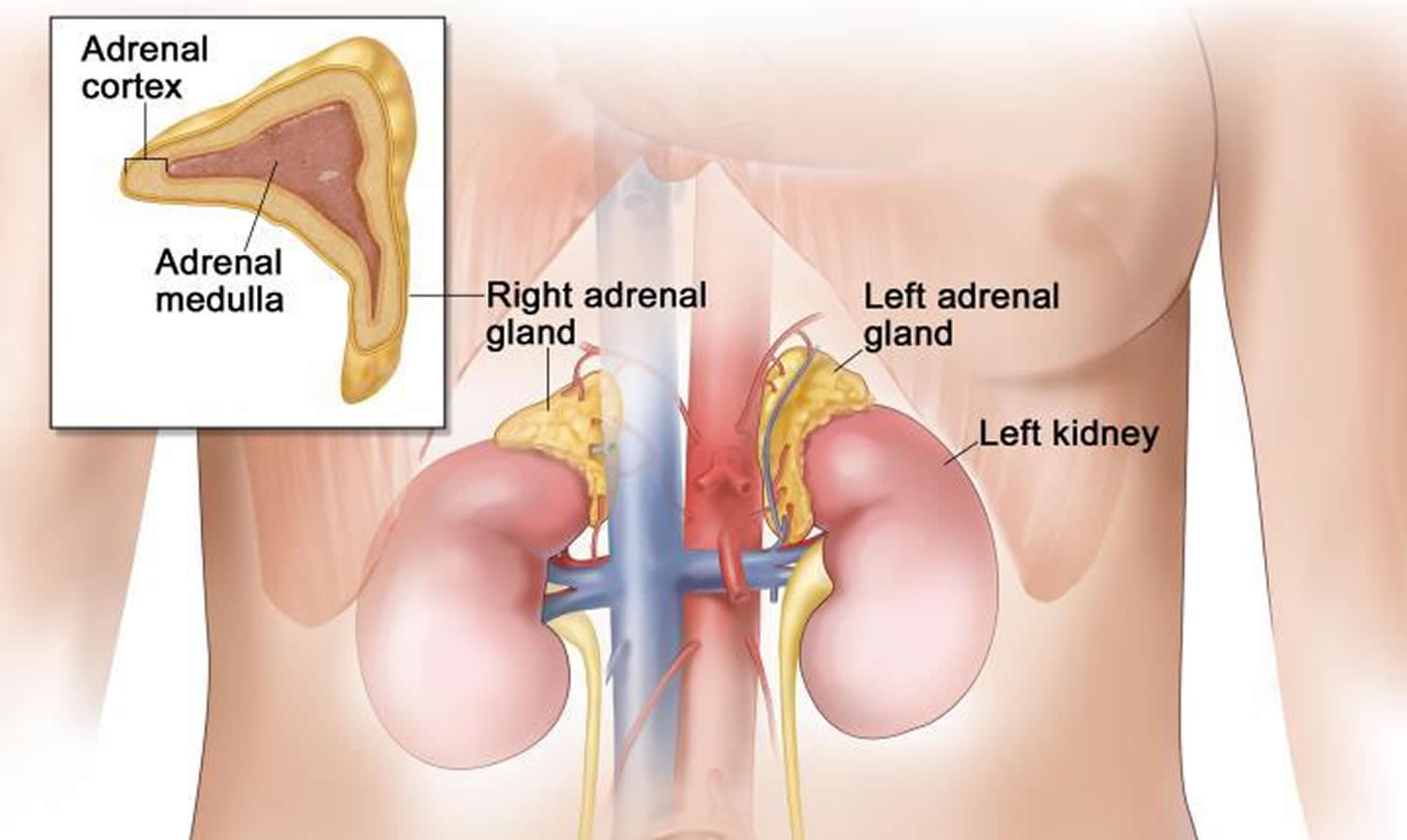
The adrenals are small glands that sit above each of the kidneys. The kidneys are located deep inside the upper part of the abdomen.
Figure 1. Adrenal gland anatomy
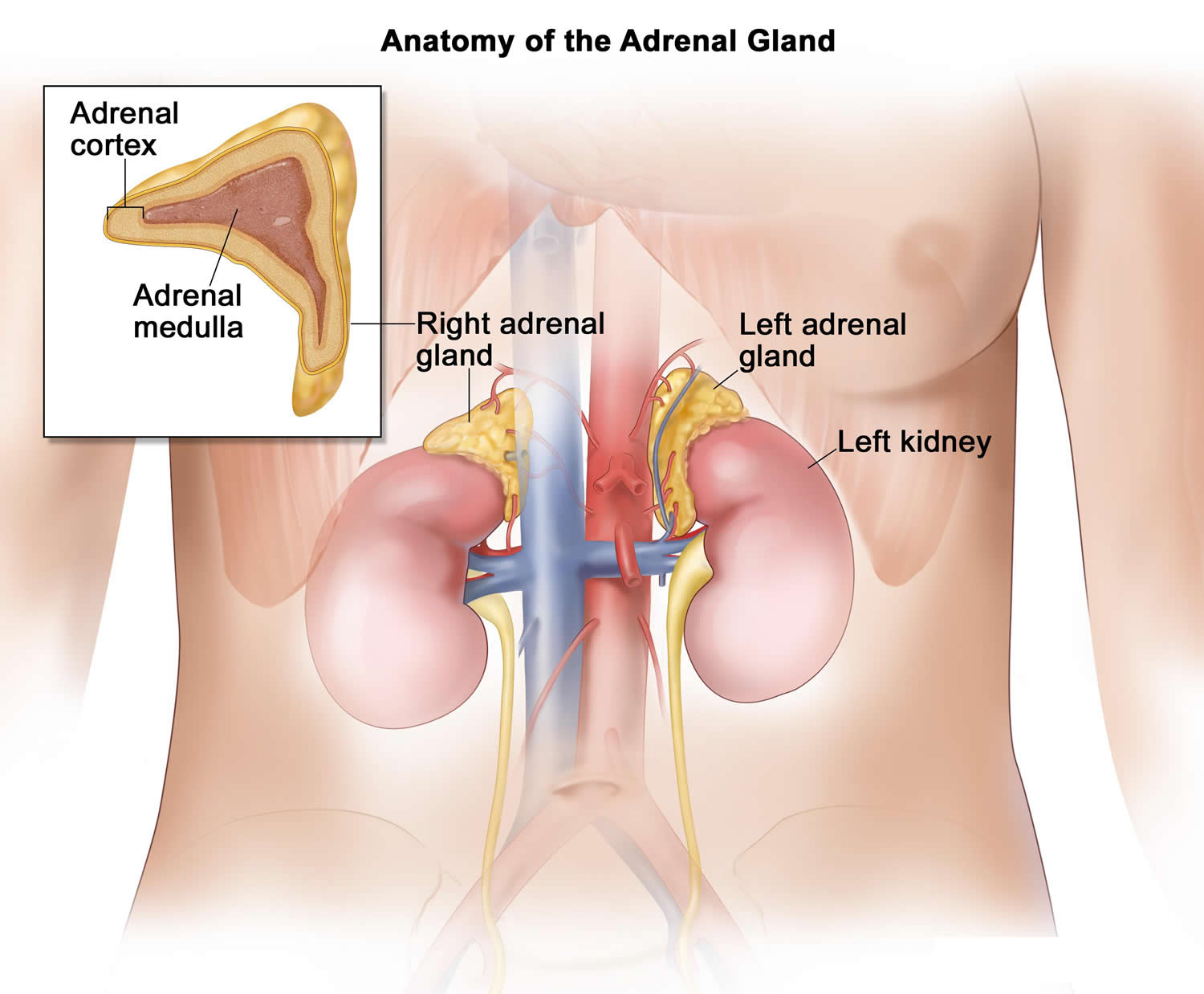
Figure 2. Adrenal gland microscopic section
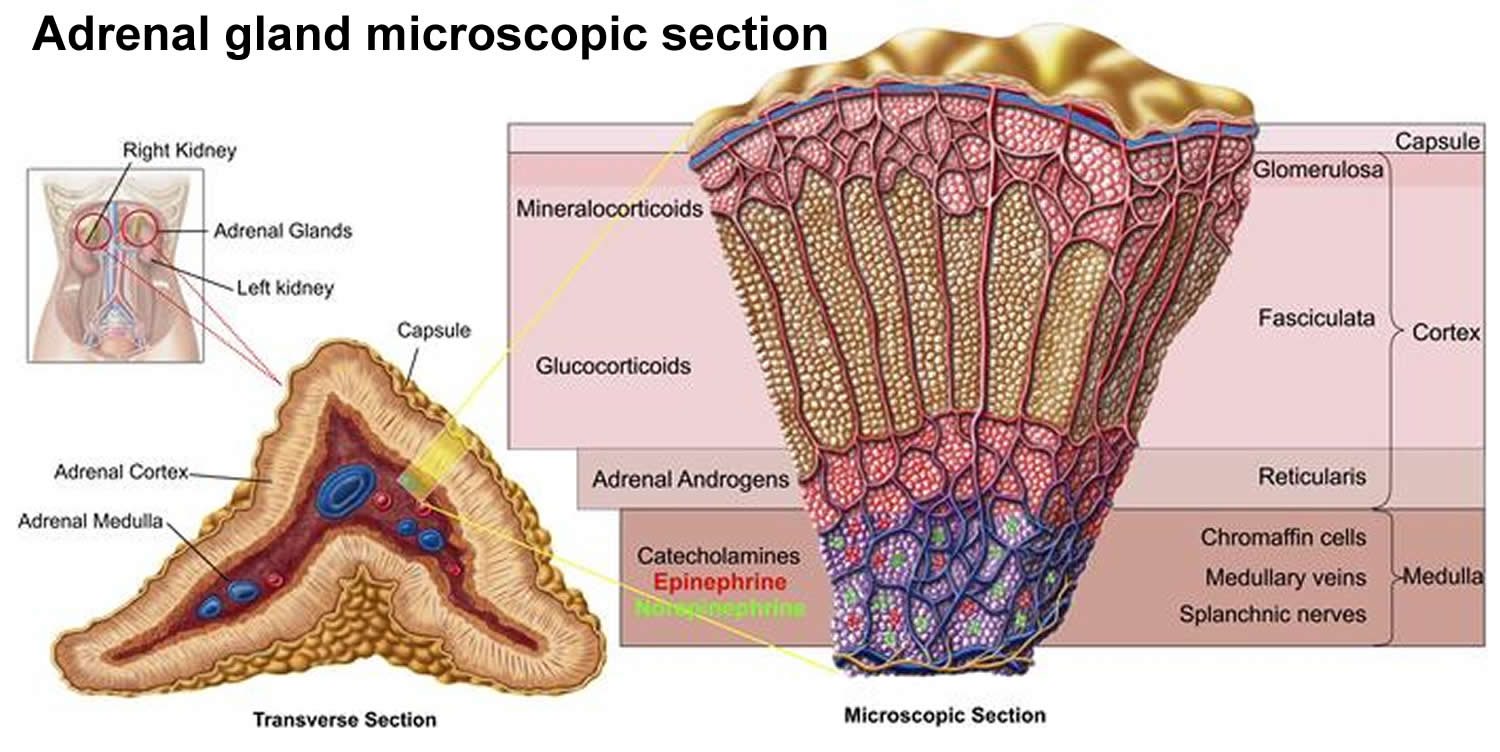
Each adrenal gland has 2 parts. The outer part, the cortex, is where most tumors develop. The cortex makes certain hormones for the body. These hormones all have a similar chemical structure and are called steroids:
- Cortisol causes changes in metabolism to help the body to handle stress.
- Aldosterone helps the kidneys regulate the amount of salt in the blood and helps regulate blood pressure.
- Adrenal androgens can be converted to more common forms of the sex hormones estrogen and testosterone in other parts of the body. The amount of these hormones that result from conversion of adrenal androgens is small compared to what is made in other parts of the body. The testicles produce most of the androgens (male hormones) in men. The ovaries produce most of the estrogens (female hormones) in women.
The inner part of the adrenal gland, the medulla, is really an extension of the nervous system. Nervous system hormones such as norepinephrine and epinephrine (also called adrenaline) are made in the medulla. Tumors and cancers that start in the adrenal medulla include pheochromocytomas (which are most often benign) and neuroblastomas.
Pheochromocytoma complications
High blood pressure can damage multiple organs, particularly tissues of the cardiovascular system, brain and kidneys. Untreated, high blood pressure associated with pheochromocytomas can result in a number of critical conditions, including:
- Heart disease
- Stroke
- Kidney failure
- Acute respiratory distress
- Damage to the nerves of the eye
Functioning pheochromocytoma create excessive amounts of catecholamines (adrenaline and noradrenaline) in your body, elevating the risk of heart attack, stroke, and even kidney failure. For patients with functioning pheochromocytoma, there is also a danger to outpatient procedures that involve anesthesia; this includes dental procedures that involve certain numbing agents. A reaction to the anesthetics causes the body to release a large amount of catecholamines that can send the patient into a catecholamine crisis and cause stroke or heart attack.
Even non-functioning pheochromocytoma tumors pose a threat by growing too large and/or causing nerve and organ damage, and because they are made of the same cells as functioning tumors, they can “switch on” at any time causing all the dangers that a functioning pheochromocytoma patient experience.
Cancerous (malignant) tumors
Rarely, a pheochromocytoma is cancerous (malignant), and the cancerous cells spread to other parts of the body (metastasize). Cancerous cells from a pheochromocytoma or paraganglioma most often migrate to the lymph system, bones, liver or lungs.
Pregnancy and pheochromocytoma/paraganglioma
Having a pheochromocytoma or paraganglioma tumor during pregnancy can be dangerous for the mother-to-be and the baby. Uncontrolled high blood pressure can damage the kidneys, restrict oxygen to the baby, or cause premature labor. During the stress of labor, a pheo or para tumor can release massive amounts of catecholamines that may cause hypertensive crisis in the mother and/or complicate the delivery. Therefore, patients with a suspected pheo/para tumor should be monitored closely during pregnancy and have their blood pressure controlled with medication. Consultation with a pheochromocytoma/paraganglioma expert is essential for the best possible outcome.
Pregnant women with pheochromocytoma need special care
Although it is rarely diagnosed during pregnancy, pheochromocytoma can be very serious for the mother and the newborn. Women who have an increased risk of pheochromocytoma should have prenatal testing. Pregnant women with pheochromocytoma should be treated by a team of doctors who are experts in this type of care.
Signs of pheochromocytoma in pregnancy may include any of the following:
- High blood pressure during the first 3 months of pregnancy.
- Sudden periods of high blood pressure.
- High blood pressure that is very hard to treat.
The diagnosis of pheochromocytoma in pregnant women includes testing for catecholamine levels in blood and urine. See the General Information section for a description of these tests and procedures. An MRI can be done to safely find tumors in pregnant women because it does not expose the fetus to radiation.
Treatment of pregnant women with pheochromocytoma may include surgery.
Treatment of pheochromocytoma during pregnancy may include the following:
- Surgery to completely remove the cancer during the second trimester (the fourth through the sixth month of pregnancy).
- Surgery to completely remove the cancer combined with delivery of the fetus by cesarean section.
Pheochromocytoma life expectancy
In most pheochromocytomas (approximately 75% to 95%), a single adrenal tumor presents itself in a patient, and the underlying cause is unknown. Once the pheochromocytoma tumor is removed, catecholamine levels stabilize and the patient can resume normal life. Patients with single, localized pheochromocytoma tumors should experience a survival rate similar to age-matched disease-free individuals. When the origin of the tumor(s) is unknown and there is no known genetic mutation in the patient, the cause is referred to as sporadic.
Of patients with initial single, localized pheochromocytoma tumors, 6.5% to 16.5% will develop a recurrence, usually 5 to 15 years after initial surgery. Although this statistic is fairly low, the consequences of missing a recurrence could be catastrophic, so continued follow-up after the first tumor is vital. In addition, it has recently been proposed that all patients diagnosed with a pheochromocytoma or paraganglioma undergo genetic testing because the incidence of a hereditary syndrome in apparently sporadic cases is as high as 25%. Early identification of a hereditary syndrome allows for early screening for other associated tumors and identification of family members who are at risk.
Although recurrent pheochromocytoma disease is rare, approximately 50% of patients with recurrent pheochromocytoma disease will experience distant metastasis. The 5-year survival in the setting of metastatic pheochromocytoma cancer (whether identified at the time of initial diagnosis or identified postoperatively as recurrent disease) is 40% to 45%. There has been debate among medical professionals about whether pheochromocytoma tumors are considered benign versus malignant; however, it is generally accepted that malignant pheochromocytoma tumors occur when tumors arise where chromaffin/glomus tissue does not normally exist in the body (such as the liver, lungs, and bones) or when there is spread of a tumor to distant sites such as distant lymph nodes or bone. Multiple tumors along the sympathetic or parasympathetic nervous system are not considered to be malignant. Malignant and multiple pheochromocytoma tumors are often associated with a genetic mutation in the patient; however, sporadically occurring disease may also be malignant and/or recurrent
Pheochromocytoma causes
Researchers don’t know what causes most (approximately 60-75%) pheochromocytoma. The tumor develops in specialized cells, called chromaffin cells, situated in the center of an adrenal gland. These cells release certain hormones, primarily adrenaline (epinephrine) and noradrenaline (norepinephrine), that help control many body functions, such as heart rate, blood pressure and blood sugar.
In approximately 25 to 40 percent of cases these tumors are the result of genetic mutations. There are several genetic syndromes that are associated with the development of pheochromocytoma and paraganglioma. The most critical and serious is multiple endocrine neoplasia (MEN) which also includes an aggressive tumor of the thyroid gland, medullary thyroid carcinoma, as well as parathyroid gland tumors (in MEN type 2A syndrome). There are other syndromes that can be associated with pheochromocytoma and an expert endocrinologist can determine what tests to perform in order to find them. The rest of pheochromocytomas are sporadic (about 60 to 75 percent) and have no genetic factors responsible for the development of the tumor. It has been suggested that all patients diagnosed with a pheochromocytoma or paraganglioma be urged to consider genetic testing because the incidence of a hereditary syndrome even in apparently sporadic cases is as high as 25-40%.
Related tumors
While most chromaffin cells reside in the adrenal glands, small clusters of these cells are also in the heart, head, neck, bladder, back wall of the abdomen and along the spine. Tumors in these chromaffin cells, called paragangliomas, may result in the same effects on the body.
Paragangliomas are closely related to pheochromocytoma. However, paras originate from the ganglia (group of nerve cells) of the nervous system, allowing them to form anywhere along the sympathetic and parasympathetic nervous systems. There are two types of paragangliomas – chromaffin paragangliomas and nonchromaffin paragangliomas.
Chromaffin paragangliomas arise from chromaffin cells, and are sometimes called extra-adrenal pheochromocytoma, which means the tumor is found outside the adrenal gland. These tumors usually secrete hormones like pheo. Chromaffin paras are rarer than pheo, but have a higher chance of malignancy (cancerous).
Nonchromaffin paragangliomas are usually named based on the primary anatomical site of origin. For example, a Carotid Body Tumor is another name for a neck paraganglioma. Nonchromaffin paras arise from glomus cells instead of chromaffin cells. They are commonly benign, and generally present in the head and neck areas. Nonchromaffin paras rarely secrete hormones, and can occur within familial lines.
Risk factors for pheochromocytoma
People with rare inherited disorders have an increased risk of developing a pheochromocytoma or paraganglioma, and tumors associated with these disorders are more likely to be cancerous. These genetic conditions include the following:
- Multiple endocrine neoplasia, type II (MEN II) is a disorder resulting in tumors in more than one part of the body’s hormone-producing (endocrine) system. Other tumors associated with MEN II can appear on the thyroid, parathyroid, lips, tongue and gastrointestinal tract.
- Von Hippel-Lindau disease can result in tumors at multiple sites, including the central nervous system, endocrine system, pancreas and kidneys.
- Neurofibromatosis 1 (NF1) results in multiple tumors in the skin (neurofibromas), pigmented skin spots and tumors of the optic nerve.
- Hereditary paraganglioma syndromes are inherited disorders that result in either pheochromocytomas or paragangliomas.
Multiple Endocrine Neoplasia Type 2
Multiple Endocrine Neoplasia, Type 2 (MEN2) is an inherited condition that is caused by genetic mutations in the RET gene on chromosome 10. When normal, these genes signal when to turn on cell growth and division. A mutation in RET causes the cell growth and division signal to always be on, which increases the risk for specific types of tumors.
Multiple Endocrine Neoplasia Type 2 (MEN2) is classified into three subtypes: MEN2A, MEN2B, and FMTC (familial medullary thyroid carcinoma). All three subtypes involve high risk for development of medullary carcinoma of the thyroid; MEN2A and MEN2B have an increased risk for pheochromocytoma (a specific type of tumor in the adrenal gland); MEN2A has an increased risk for parathyroid adenoma or hyperplasia (excessive growth). Additional features in MEN2B include bumps (neuromas) of the lips and tongue; enlarged lips; and ganglioneuromas (a specific type of polyp within the gastrointestinal tract),. In addition, patient’s with MEN2B tend to be slender with long limbs. About 5% of MEN2A patients and 50% of MEN2B patients have the disease because of a de novo (new) mutation that was not inherited from their parents. If an individual has a RET mutation, then each of his or her children will have a 50% chance of having MEN2, as well.
Neurofibromatosis Type 1 (NF1)
Neurofibromatosis Type 1 (NF1) is an inherited condition caused by genetic mutations in the NF-1 gene on chromosome 17. When normal, these genes help stop tumors from developing. A mutation in NF-1 increases the risk for multiple café au lait spots; axillary and inguinal freckling; multiple cutaneous (skin) neurofibromas; and iris Lisch nodules. Learning disabilities are present in at least 50% of individuals with NF1. Less common but potentially more serious manifestations include plexiform neurofibromas; optic nerve and other central nervous system gliomas; malignant peripheral nerve sheath tumors; scoliosis; tibial dysplasia; and vasculopathy.
If an individual has an NF-1 mutation, each of his or her children will have a 50% chance of having Neurofibromatosis Type 1 (NF1) as well.
Von-Hippel Lindau Disease
Von-Hippel Lindau is an inherited condition caused by genetic mutations in the VHL gene on chromosome 3. When normal, this gene helps stop tumors from developing. A mutation in the VHL gene increases the risk for many types of benign and cancerous tumors in the brain, spinal cord, eye, ear, kidneys, adrenal glands, and other parts of the body. If an individual has a Von-Hippel Lindau mutation, each of his or her children will have a 50% chance of having Von-Hippel Lindau as well. Over 90% of patients with this genetic mutation will develop disease by the age of 65. Approximately 20% of Von-Hippel Lindau patients will develop pheochromocytoma. The severity of symptoms varies widely between individuals.
Hereditary Paraganglioma Syndrome
Hereditary Paraganglioma Syndrome is an inherited condition caused by genetic mutations in the SDHD, SDHB, SDHC, SDHA, and SDHAF2 (also known as SDH5) genes. SDHA, SDHB, SDHC and SDHD are four nuclear genes that encode the four subunits of the mitochondrial enzyme succinate dehydrogenase. A fifth nuclear gene, SDHAF2 (SDH5) encodes a protein that appears to be required for flavination of another succinate dehydrogenase subunit, SDHA. These are collectively known as the SDHx genes. Pathogenic variants in MAX predispose to pheochromocytoma; a subset of individuals with pathogenic variants in MAX will also develop PGL. KIF1B, EGLN1 (formerly known as PHD2), IDH1, and FH have been reported to be associated with hereditary paraganglioma/pheochromocytoma, but their clinical significance is still unclear.When normal, these genes help stop tumors from developing. A mutation in one of these genes increases the risk for paragangliomas (tumors that occur in nervous and endocrine tissues) and pheochromocytomas (paragangliomas on the adrenal gland, which is on top of the kidney). This disorder can present due to de novo (new) mutations; therefore, the absence of a family history of paraganglioma, pheochromocytoma or other features of these syndromes does not eliminate the chance that an individual has a hereditary syndrome. Other tumor types can also occur.
If an individual has an succinate dehydrogenase mutation, each of his or her children will have a 50% chance of having the same mutation. Individuals with mutations in SDHD only may develop pheo/para if they inherit the mutation from their father. Individuals with mutations in SDHB or SDHC may develop pheo/para regardless of which parent they inherit the mutation from. A single mutation in the succinate dehydrogenase genes increases the risk that an individual will develop the disease associated with it. However, an additional mutation that deletes the normal copy of the gene is needed to cause tumor formation. This second mutation, called a somatic mutation, is acquired during a person’s lifetime and is present only in tumor cells.
Other genetic causes of pheochromocytoma and paraganglioma are being studied. For example, germline mutations in the gene TMEM127 on chromosome 2q11 have been shown to be present in approximately 30% of affected patients with familial disease and in about 3% of patients with apparently sporadic pheochromocytomas without a known genetic cause. Similar to the SDH genes, TMEM127 is a tumor-suppressing gene.
More recently, the TMEM127, MAX, HIF2A, EGLN1, KIF1B, and H-RAS were added to the list of susceptibility genes implicated in the development of paragangliomas/pheochromocytomas. Researchers are still studying the hereditary patterns and penetrance of these mutations.
Carney Triad and Carney-Stratakis Diad
Carney Triad is a rare disease that causes three different tumor types to develop: functioning paragangliomas, pulmonary chondromas (benign cartilaginous lung tumors), and GISTs (gastrointestinal stromal tumors). gastrointestinal stromal tumors may occur anywhere inside the digestive tract, but the stomach is the most common area; they may be multifocal. Carney Triad affects more women than men; up to 80% of patients are women. Even though a gene mutation has not been discovered, it is strongly suspected that Carney Triad is genetic.
Carney-Stratakis Diad is similar to Carney Triad; however, gene mutations causing this condition have been identified. Patients with the condition have been found to have mutations in the SDHB, SDHC, and SDHD genes. The condition is characterized by paraganglioma tumors and gastrointestinal stromal tumor (no pulmonary chondromas as with Carney Triad). Carney-Stratakis Diad is autosomal dominant.
Pheochromocytoma symptoms
The most common sign of pheochromocytoma is high blood pressure which is sometimes sudden and extreme. Symptoms may include anxiety or emotional stress, rapid pulse, palpitations, headache, nausea, vomiting and clammy skin. Recurrence or intensification of Significant elevation of blood pressure may lead to a heart attack or stroke. The person may experience abdominal or back pressure, although rarely pain, from the growing tumor. There is a wide variation in symptoms in affected persons.
Signs and symptoms of pheochromocytomas often include:
- High blood pressure
- Heavy sweating
- Sudden-onset of headaches (usually severe)
- Rapid heartbeat (tachycardia) / forceful heartbeat, even while resting
- Tremors
- Paleness in the face (pallor)
- Shortness of breath (dyspnea)
- Flushing
- Chest or abdominal pain
Less common signs or symptoms may include:
- Attacks similar to panic/anxiety attacks (these may occur in response to exercise or for no apparent reason)
- Anxiety or sense of doom
- Constipation
- Weight loss
- Emotional mood swings
- Joint pains
- Pale Skin
Triggers of symptomatic spells
Spells may occur spontaneously or may be triggered by such factors as:
- Physical exertion
- Anxiety or stress
- Changes in body position
- Labor and delivery
- Surgery and anesthesia
Pheochromocytoma diet
Foods high in tyramine, a substance that affects blood pressure, also can trigger a spell. Tyramine is common in foods that are fermented, aged, pickled, cured, overripe or spoiled. These foods include:
- Some cheeses
- Some beers and wines
- Chocolate
- Dried or smoked meats
Certain medications that can trigger a symptomatic spell include:
- Monoamine oxidase inhibitors (MAOIs), such as phenelzine (Nardil), tranylcypromine (Parnate) and isocarboxazid (Marplan)
- Stimulants, such as amphetamines or cocaine
Pheochromocytoma symptoms may occur as intermittent episodes rather than a persistent progression of the disease. These attacks or symptoms often occur during “episodes” which typically last 20 to 60 minutes and are not sustained; however 50% to 60% of pheochromocytoma patients do have sustained hypertension. These symptoms can include dramatic elevations of blood pressure that occur spontaneously or could be induced. Direct trauma, mechanical pressure to the tumor or any type of stress can precipitate hypertensive episodes. These may include anesthesia, surgery, exercise, defecation, sexual intercourse, pregnancy, parturition, alcohol consumption, smoking, administration of various medications or urination (if the bladder wall is involved).
Pheochromocytoma diagnosis
In addition to a complete medical history, physical examination, and family history, diagnostic procedures for pheochromocytoma include:
Pheochromocytoma test
- Blood and urine tests to measure hormone levels such as 24-hour urinary catecholamines and metanephrines, serum catecholamines, metanephrines and chromogranin A levels.
- Twenty-four-hour urine collection: This test measures catecholamine levels (e.g., epinephrine, norepinephrine, and dopamine) as well as fractionated metanaphrines (e.g., metanephrine and normetanephrine) in the urine. Patients will collect their urine for 24 hours and return it to the lab. Typically a preservative is used and/or the sample is kept cold during the testing period.
- Plasma-free fractionated metanephrines: This test measures fractionated metanaphrine levels in the blood. For best results, the patient should lie supine for 30 minutes before the blood draw.
The 24-hour urine collection has a relatively low sensitivity (77-90%) which means it can miss the diagnosis approximately 10-25% of the time and therefore may need to be repeated if there is a strong suspicion of pheochromocytoma/paraganglioma but the first test results come back negative. The sensitivity for this test is however very good (98%), which means there are very few false positives. In contrast, the sensitivity of the plasma-free fractionated metanephrines test is very good (97-99%) but a relatively low specificity (85%), so false positives are fairly common. Ideally both tests should be used to confirm or refute the pheo diagnosis. The plasma-free fractionated metanephrines test should be done first to increase certainty of the tumor’s presence, followed by the 24-hour urine test for confirmation.
Certain foods and drugs may affect the outcome of these tests. Patients should avoid caffeine, bananas, chocolate, and acetaminophen (Tylenol) for at least 24 hours before the test (and during the collection). In addition, stress and sleep apnea can affect the results.
A mildly elevated catecholamine or metanephrine level is usually the result of drug interactions or other factors. Patients with symptomatic pheo/para almost always have increases in catecholamines or metanephrines of at least two to three times higher than the upper limits of reference ranges.
If the catecholamine levels measured in these tests are elevated, the next step is to order scans to localize the tumor(s). CT (computed tomography) or MRI (magnetic resonance imaging) of the pelvis and abdomen (at least up to the bifurcation of the aorta) are generally accepted as the first set of scans used to locate pheochromocytoma.
Imaging tests
If the results of laboratory tests indicate the possibility of a pheochromocytoma or paraganglioma, your doctor will order one or more imaging tests to locate a possible tumor. These tests may include:
- CT scan of the abdomen.
- MRI of the abdomen.
- MIBG (iodine-131-meta-iodobenzylguanidine) scan. The MIBG scan is a functional study utilizing a radioisotope that produces an image of the functioning adrenal gland or extra- adrenal paraganglioma.
- Positron emission tomography (PET), a scanning technology that can also detect radioactive compounds taken up by a tumor.
Both Ct scan and MRI have similar sensitivities (90-100%) and specificities (70-80%). Additional imaging may be necessary if CT and MRI fail to localize the tumor. Several types of scans are used to detect pheochromocytoma and paraganglioma tumors, including (131)I-MIBG, (123)I-MIBG, (111)In-octreotide and (18)F-flurodeoxyglugose PET scans. MIBG, octreotide and PET scans are special types of scans that involve injecting the patient with a radioactive tracer. Pheo and para tumors will absorb the tracer and light up on the scan if they have the receptors for the tracers on their cell walls. Please note that not all pheo/para tumors show up on MIBG and octreotide scans. Some tumors don’t take up the radioactive tracers.
Incidental discovery
A tumor in an adrenal gland might be found during imaging studies conducted for other reasons. In such cases, your doctor would order additional tests to determine the nature of the tumor.
Genetic testing
Your doctor might recommend genetic tests to determine if a pheochromocytoma is related to an inherited disorder. Information about possible genetic factors can be important for a number of reasons:
- Because some inherited disorders can cause multiple conditions, test results may indicate the need to screen for other medical problems.
- Because some disorders are more likely to be recurrent or cancerous (malignant), your test results may affect treatment decisions or long-term plans to monitor your health.
- Results from your tests may indicate that other family members should be screened for pheochromocytoma or related conditions.
Ask your doctor about genetic counseling services that can help you understand the benefits and implications of genetic testing.
Pheochromocytoma treatment
The primary treatment for a pheochromocytoma is surgery to remove the tumor, even in cases where there are pheochromocytomas in both adrenal glands 6. Prior to surgery, it is important to take medications to control and stabilize blood pressure. Before you undergo surgery, your doctor will prescribe specific blood pressure medications that block the actions of the high-adrenaline hormones to lower the risk of developing dangerously high blood pressure during surgery. Following surgery, affected individuals should visit their physicians regularly to monitor blood pressure and blood levels of metanephrine 7. Long-term treatment with medication should only be considered if, for some reason, the pheochromocytoma cannot be removed by surgery 8.
Pre-operative medications
You will likely take two drugs for seven to 10 days that help lower blood pressure before surgery. These drugs will either replace or be added to other blood pressure drugs you take.
- Alpha blockers keep smaller arteries and veins open and relaxed, improving blood flow and decreasing blood pressure. Alpha blockers include phenoxybenzamine (Dibenzyline), doxazosin (Cardura), and prazosin (Minipress). Side effects might include irregular heartbeat, dizziness, fatigue, vision problems, sexual dysfunction in men and swelling in your limbs.
- Beta blockers cause your heart to beat more slowly and with less force. They also help keep blood vessels open and relaxed. In preparing for surgery a beta blocker is added several days after starting the alpha blocker. Beta blockers include atenolol (Tenormin), metoprolol (Lopressor, Toprol-XL) and propranolol (Inderal, Innopran XL). Possible side effects include fatigue, upset stomach, headache, dizziness, constipation, diarrhea, irregular heartbeat, difficulty breathing and swelling in the limbs.
- High-salt diet. Alpha and beta blockers widen (dilate) the blood vessels, causing the amount of fluid within the blood vessels to be low. This can cause dangerous drops in blood pressure with standing. A high-salt diet will draw more fluid inside the blood vessels, preventing the development of low blood pressure during and after surgery.
Surgery
In most cases, the entire adrenal gland with a pheochromocytoma is removed with laparoscopic, or minimally invasive, surgery. Your surgeon will make a few small openings through which he or she inserts wandlike devices equipped with video cameras and small tools.
The remaining healthy adrenal gland carries out the functions normally performed by two, and blood pressure usually returns to normal.
In some unusual situations, such as when the other adrenal gland has been removed, surgery might remove only the tumor, sparing some healthy tissue.
If a tumor is cancerous (malignant), the effectiveness of surgery might rely on removing the tumor and all cancerous tissue. However, even if all of the cancerous tissue isn’t removed, surgery might limit hormone production and provide some control of blood pressure.
Cancer treatments
As cancer is rare among cases of pheochromocytoma, the research about the best treatments is relatively limited. Treatments for malignant tumors and metastasized cancer related to a pheochromocytoma might include:
- Radionuclide treatment. This radiation therapy combines MIBG, a compound that attaches to adrenal tumors, with a type of radioactive iodine. The treatment goal is to deliver radiation therapy to a specific site and kill cancerous cells.
- Chemotherapy. Chemotherapy is the use of powerful drugs that kill fast-growing cancer cells.
- Targeted cancer therapies. These medications inhibit the function of naturally occurring molecules that promote the growth and spread of cancerous cells.
Radiotherapy
External beam radiation has been successful in treating a variety of cancers, but it has mixed results with pheo/para tumors. It can be effective as a method of pain control, but it may not eliminate the tumor cells. Side effects include fatigue, skin burns at the site of radiation, nerve damage, and arthritis (long term).
Radio Frequency Ablation
Radio Frequency Ablation is a relatively new procedure that involves inserting a needle directly into the tumor and destroying the cells with radio waves. This procedure is not effective on large tumors. Radio Frequency Ablation should not be used on tumors in the head and neck or near nerves because of potential damage to surrounding structures. As with surgery, the patient must be blocked with an alpha blocker for at least 2-3 weeks before the procedure. Side effects include pain and swelling at the site of the procedure.
Transarterial embolization
Transarterial embolization is used for liver tumors that are too large for radio frequency ablation (typically over 5cm). In this procedure, a catheter (a thin, flexible tube) is put into an artery through a small cut in the inner thigh and threaded up into the hepatic artery in the liver. A dye is usually injected into the bloodstream at this time to help the doctor monitor the path of the catheter via angiography. Once the catheter is in place, small particles are injected into the artery to plug it up and cut off the blood supply.
MIBG (I-131 metaiodobenzylguanidine)
This is the same radioactive tracer that is used during scans to locate tumors, but in a much higher dose. In order to receive MIBG (I-131 metaiodobenzylguanidine) as a therapy, the pheo/para tumors must be MIBG (I-131 metaiodobenzylguanidine) positive, meaning they will absorb, or “take up,” the tracer. Precautions must be taken to protect the patient’s thyroid. Side effects of this treatment include fatigue, nausea, and a decrease in blood platelets, especially in high-dose treatments or with several treatments over time.
Radiolabeled Somatostatin Analogues
Symptomatic improvement may occur with all of the various (111)In, (90)Y, or (177)Lu-labelled somatostatin analogues that have been used. Since tumor size reduction was seldom achieved with (111)Indium labelled somatostatin analogues, radiolabelled somatostatin analogues with beta-emitting isotopes like (90)Y and (177)Lu were developed. Reported anti-tumor effects vary considerably between various studies. The side effects are few and mostly mild from these treatments. However, like MIBG (I-131 metaiodobenzylguanidine), the tumors must be receptive to the agents being considered for use. Not all patients will be candidates for these therapies because their tumor cells won’t “take up” the tracer elements.
Octreotide
Octreotide is a synthetic octapeptide that mimics somatostatin but is a more potent inhibitor of growth hormone, glucagon, and insulin than natural somatostatins. Like the radio labeled somatostatin analogues, Octreotide can also be labelled with a variety of radionuclides, such as yttrium-90 or lutetium-177, to enable peptide receptor radionuclide therapy for the treatment of unresectable pheochromocytoma/paraganglioma.
Chemotherapy
The type of chemotherapy used for pheo/para tumors is a mix of three drugs: Cyclophosphamide (Cytoxan), Vincristine, and Dacarbozine (DTIC). This type of chemo is referred to as CVD. Typically, patients will remain on CVD for an extended period of time. A common error with this therapy is to stop the therapy too soon, resulting in further tumor growth. Once CVD chemotherapy has been stopped it typically cannot be started again. It is recommended that patients stay on chemo for 20 or more rounds. An experienced physician should manage this protocol. Patients with the SDHB mutation in particular seem to respond well to this treatment. Side effects include fatigue, nausea, hair loss, decreased white and red blood cell counts, and low platelets. If the patient has bone metastases, a bone-strengthening drug (bisphosphonate) may also be administered with the chemo.
Molecular Targeted Therapy
Understanding the molecular pathway changes responsible for malignant paras will hopefully guide future molecular-targeted therapies. These therapies work by interfering with specific molecular targets along the signaling pathways in the cell that are responsible for carcinogenesis and tumor growth. To date, both benign and malignant PGLs gene mutations are part of two distinct molecular pathways leading to tumorigenesis:
- Cluster 1 includes mutations of VHL, SDHB, and SDHD and is associated with pseudohypoxia and aberrant VEGF signaling, leading to abnormal hypoxia inducible factor (HIF) activation and overexpression of angiogenic factors.
- Cluster 2 includes mutations of RET, NF1, TMEM127, and MAX and is associated with abnormal activation of kinase-signaling pathways such as PI3kinase/AKT, RAS/RAF/ERK, and mTOR1/p70s6K, leading to abnormal cell growth and lack of apoptosis capacity.
This all sounds really complex, and it is, but what researchers are attempting to do is keep these cells from malfunctioning by interfering with their abnormal activities somewhere along these pathways.
HIF1a inihbitors are drugs targeted at interfering with HIF hypoxia-driven transcription pathway. These agents have shown marked anti-tumor activity in mice models and seem to be promising for malignant paras, but more studies are needed.
The mTOR inhibitor everolimus (RAD001) in combination with octreotide LAR has been evaluated for low- and intermediate-grade neuroendocrine tumors, with good results. However, when everolimus was evaluated in malignant para patients, all patients experienced disease progression. Researchers concluded that further studies on the PI3K/AKT/mTOR pathway have to be conducted to find a more specific molecular target in its signalling.
Several studies have demonstrated overexpression in malignant pheo/para of angiogenic molecules, such as VEGF, angiopoietin-2, and the endothelin receptors ETA and ETB, suggesting that targeting this pathway with antiangiogenic therapies could represent a new promising treatment option. As a result, sunitinib, a receptor tyrosine kinase inhibitor that acts on several targets (VEGF, PDGF, and c-KIT), and has antiangiogenic and antitumor activity, has been used in the treatment of malignant pheo/para, with mixed but promising results.
Imatinib, another tyrosine kinase inhibitor already used for hematologic and gastrointestinal stromal tumors, has not been found effective for malignant pheochromocytoma/paraganglioma treatment.
Thalidomide, by targeting VEGF and basic fibroblast growth factor, is an antiangiogenic agent that has been evaluated in combination with Temozolomide in neuroendocrine tumors. Although there was an objective biochemical response rate (40%) and a radiologic response rate in 33% of malignant pheo/paras, lymphopenia occurred in about 70% of treated patients.
Activators of prolyl hydroxylase (PHD) (such as ERBB2 inhibitors) are now being evaluated as promising antineoplastic therapies. These molecules decrease the expression levels of some angiogenic factors, such as VEGF, acting on HIF pathway, by activating the PHD, thus increasing HIF hydroxylation, and promoting its degradation. More studies are required on these agents.
Molecular targeted therapies are promising strategies, but, due to the complexity of the tumor pathogenesis, further studies on tumor biology, discovery of novel targeted drugs, and new trials are needed to achieve more effective treatments.
Follow-up tests will be needed
Some of the tests that were done to diagnose the cancer or to find out the extent of the cancer may be repeated. Some tests will be repeated in order to see how well the treatment is working. Decisions about whether to continue, change, or stop treatment will be based on the results of these tests.
Some of the tests will continue to be done from time to time after treatment has ended. The results of these tests can show if your condition has changed or if the cancer has recurred (come back). These tests are sometimes called follow-up tests.
For patients with pheochromocytoma or paraganglioma that causes symptoms, catecholamine levels in the blood and urine will be checked on a regular basis. Catecholamine levels that are higher than normal can be a sign that the cancer has come back.
For patients with paraganglioma that does not cause symptoms, follow-up tests such as CT, MRI, or MIBG scan should be done every year.
For patients with inherited pheochromocytoma, catecholamine levels in the blood and urine will be checked on a regular basis. Other screening tests will be done to check for other tumors that are linked to the inherited syndrome.
Talk to your doctor about which tests should be done and how often. Patients with pheochromocytoma or paraganglioma need lifelong follow-up.
- Kirmani S, Young WF. Hereditary Paraganglioma-Pheochromocytoma Syndromes. 2008 May 21 [Updated 2014 Nov 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1548/[↩]
- CARNEY TRIAD. http://www.omim.org/entry/604287[↩]
- Carney, J. A., Sheps, S. G., Go, V. L. W., Gordon, H. The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. New Eng. J. Med. 296: 1517-1518, 1977. http://www.nejm.org/doi/full/10.1056/NEJM197706302962609[↩]
- Stratakis CA. New genes and/or molecular pathways associated with adrenal hyperplasias and related adrenocortical tumors. Mol Cell Endocrinol. 2009;300:152–7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2668239/[↩][↩]
- Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74:543–52. https://www.ncbi.nlm.nih.gov/pubmed/10377927[↩]
- Gumbs AA, Gagner M. Laparoscopic adrenalectomy. Best Practice & Research Clinical Endocrinology & Metabolism. 2006; 20(3):483-499. https://www.ncbi.nlm.nih.gov/pubmed/16980207[↩]
- National Comprehensive Cancer Network. Neuroendocrine Tumors. NCCN Clinical Practice Guidelines in Oncology. 2014 https://www.nccn.org/[↩]
- http://emedicine.medscape.com/article/124059-overview[↩]





{kind=link}