What is pseudocholinesterase deficiency
Pseudocholinesterase deficiency is a condition that results in increased sensitivity to certain muscle relaxant drugs used during general anesthesia, called choline esters, such as those found in succinylcholine, mivacurium, procaine, chloroprocaine, tetracaine, cocaine and heroine 1. These fast-acting drugs, such as succinylcholine and mivacurium, are given to relax the muscles used for movement (skeletal muscles), including the muscles involved in breathing 2. The drugs are often employed for brief surgical procedures or in emergencies when a breathing tube must be inserted quickly. Normally, these drugs are broken down (metabolized) by the body within a few minutes of being administered, at which time the muscles can move again. However, people with pseudocholinesterase deficiency may not be able to move or breathe on their own for a few hours after the drugs are administered. Affected individuals must be supported with a machine to help them breathe (mechanical ventilation) until the drugs are cleared from the body.
People with pseudocholinesterase (also known as butyrylcholinesterase) deficiency may also have increased sensitivity to certain other drugs, including the local anesthetic procaine, and to specific agricultural pesticides. Pseudocholinesterase also helps protect the body by breaking down certain toxic substances before they reach the nerves. These substances include certain pesticides, poisons that attack the nerves, and specific natural toxins including a compound called solanine found in green potato skin. It is likely that the enzyme has other functions in the body, but these functions are not well understood. Studies suggest that the enzyme may be involved in the transmission of nerve signals.
Pseudocholinesterase deficiency causes no other signs or symptoms and is usually not discovered until an abnormal drug reaction occurs.
Pseudocholinesterase deficiency occurs in 1 in 3,200 to 1 in 5,000 people. It is more common in certain populations, such as the Persian Jewish community and Alaska Natives. Pseudocholinesterase deficiency can be inherited (genetic) or acquired. When it is inherited, it is autosomal recessive and caused by mutations in the BCHE gene. Acquired pseudocholinesterase deficiency may have various causes such as chronic infection, kidney or liver disease, malnutrition, major burns, cancer, or various medications 3.
Pseudocholinesterase is a tetrameric glycoprotein enzyme with a complex molecular structure 4. Pseudocholinesterase is synthesized in the liver and immediately released into the plasma 5. The plasma half-life has been estimated to be approximately 12 days 6. Deficiency or reduced activity of pseudocholinesterase enzyme results in significant prolongation of mivacurium or succinylcholine induced neuromuscular blockade 7. In addition, pseudocholinesterase activity may be reduced by a number of disease states or by concomitant drug administration.
Mivacurium, which is a nondepolarizing neuromuscular blocking drug administered in doses of 0.1 to 0.2 mg/kg, also produces rapid onset of neuromuscular blockade lasting 15 to 30 minutes 8. The rapid ester hydrolysis of mivacurium by pseudocholinesterase results in the short duration of action of this drug, which is ideal for providing muscle relaxation for brief surgical procedures 9. But, the duration of mivacurium in adults is inversely related to serum pseudocholinesterase activity 10.
Other names for pseudocholinesterase deficiency
- butyrylcholinesterase deficiency
- cholinesterase II deficiency
- deficiency of butyrylcholine esterase
- pseudocholinesterase E1 deficiency
- succinylcholine sensitivity
- suxamethonium sensitivity
Pseudocholinesterase deficiency causes
Pseudocholinesterase deficiency can be caused by mutations in the BCHE gene. This gene provides instructions for making the pseudocholinesterase enzyme, also known as butyrylcholinesterase, which is produced by the liver and circulates in the blood. The pseudocholinesterase enzyme is involved in the breakdown of choline ester drugs. It is likely that the enzyme has other functions in the body, but these functions are not well understood. Studies suggest that the enzyme may be involved in the transmission of nerve signals.
The BCHE gene that codes for the pseudocholinesterase enzyme is located at the E1 locus on the long arm of chromosome 3 and 96% of the population is homozygous for the normal pseudocholinesterase genotype, which is designated as EuEu. The remaining 4% of the population carry one or more of the following atypical gene alleles (See Table 1, below) for the pseudocholinesterase gene in either a heterozygous or homozygous fashion.
Table 1. Atypical Gene Alleles for the Pseudocholinesterase Genotype
| Ea | Atypical dibucaine-resistant variant | Point mutation |
| Ef | Fluoride-resistant variant | Point mutation |
| Es | Silent variant | Frameshift mutation |
Note: *These alleles may occur either in the homozygous form or in any heterozygous combination with each other, with the normal Eu allele, or with a number of additional rare variant abnormal alleles.
Some BCHE gene mutations that cause pseudocholinesterase deficiency result in an abnormal pseudocholinesterase enzyme that does not function properly. Other mutations prevent the production of the pseudocholinesterase enzyme. A lack of functional pseudocholinesterase enzyme impairs the body’s ability to break down choline ester drugs efficiently, leading to abnormally prolonged drug effects.
In individuals with an inherited form of pseudocholinesterase deficiency, only a single atypical allele is carried in a heterozygous fashion, resulting in a partial deficiency in enzyme activity, which manifests as a slightly prolonged duration of paralysis, longer than 5 minutes but shorter than 1 hour, following administration of succinylcholine. The heterozygous atypical form affects anywhere from 1 in 25, to 1 in 480 individuals (depending on the severity of the condition) 1. Less than 0.1% of the general population carries 2 pseudocholinesterase gene allele (homozygous) mutations that will produce clinically significant effects from succinylcholine lasting longer than 1 hour.
One rare variant allele of the pseudocholinesterase gene, designated the C5 variant, actually has higher than normal enzyme activity, resulting in relative resistance to the paralytic effects of succinylcholine.
The dibucaine-resistant genetic variant form of pseudocholinesterase is identified by the percent inhibition of hydrolysis of benzyl choline caused by the addition of dibucaine to the pseudocholinesterase enzymatic assay. The dibucaine number is the percent inhibition of hydrolysis of benzyl choline by dibucaine added to the plasma sample. The normal dibucaine number for the homozygous typical genotype (EuEu) is 80%. Individuals homozygous for the atypical dibucaine resistant genotype (EaEa) have a dibucaine number of 20%, which correlates with a marked prolongation of the paralytic effect of standard doses of succinylcholine to well over 1-hour duration. Heterozygotes (EuEa) have intermediate dibucaine numbers and modest prolongation of muscle paralysis with succinylcholine. The EuEa heterozygous genotype is found in 2.5% of the general population, making it more common than all other abnormal pseudocholinesterase genotypes combined.
The fluoride-resistant pseudocholinesterase enzyme variant is identified by its percent inhibition of benzyl choline hydrolysis when fluoride is added to the assay. The fluoride number (percentage inhibition of enzyme activity in the presence of fluoride) is 60% for the EuEu genotype and is 36% for the EfEf genotype. This homozygous fluoride-resistant genotype exhibits mild to moderate prolongation of succinylcholine-induced paralysis. The heterozygous fluoride-resistant genotype usually is clinically insignificant unless accompanied by a second abnormal allele or by a coexisting acquired cause of pseudocholinesterase deficiency.
The most severe form of inherited pseudocholinesterase deficiency occurs in only 1 in 100,000 individuals who are homozygous for the silent Es genotype, with no detectible pseudocholinesterase enzyme activity. These individuals may exhibit prolonged muscle paralysis for as long as 8 hours following a single dose of succinylcholine. Gene mutations that produce silent alleles are caused by frameshift or stop codon mutations, resulting in no functional pseudocholinesterase enzyme synthesis.
Prolonged paralysis due to pseudocholinesterase deficiency has been reported after succinylcholine administration for emergent cesarean section. Abnormal pseudocholinesterase enzyme variants can be present that are undetectable with standard laboratory tests 11.
Pseudocholinesterase deficiency can also have nongenetic causes. In these cases, the condition is called acquired pseudocholinesterase deficiency; it is not inherited and cannot be passed to the next generation. Activity of the pseudocholinesterase enzyme can be impaired by kidney or liver disease, malnutrition, major burns, cancer, or certain drugs.
Pseudocholinesterase deficiency carrier
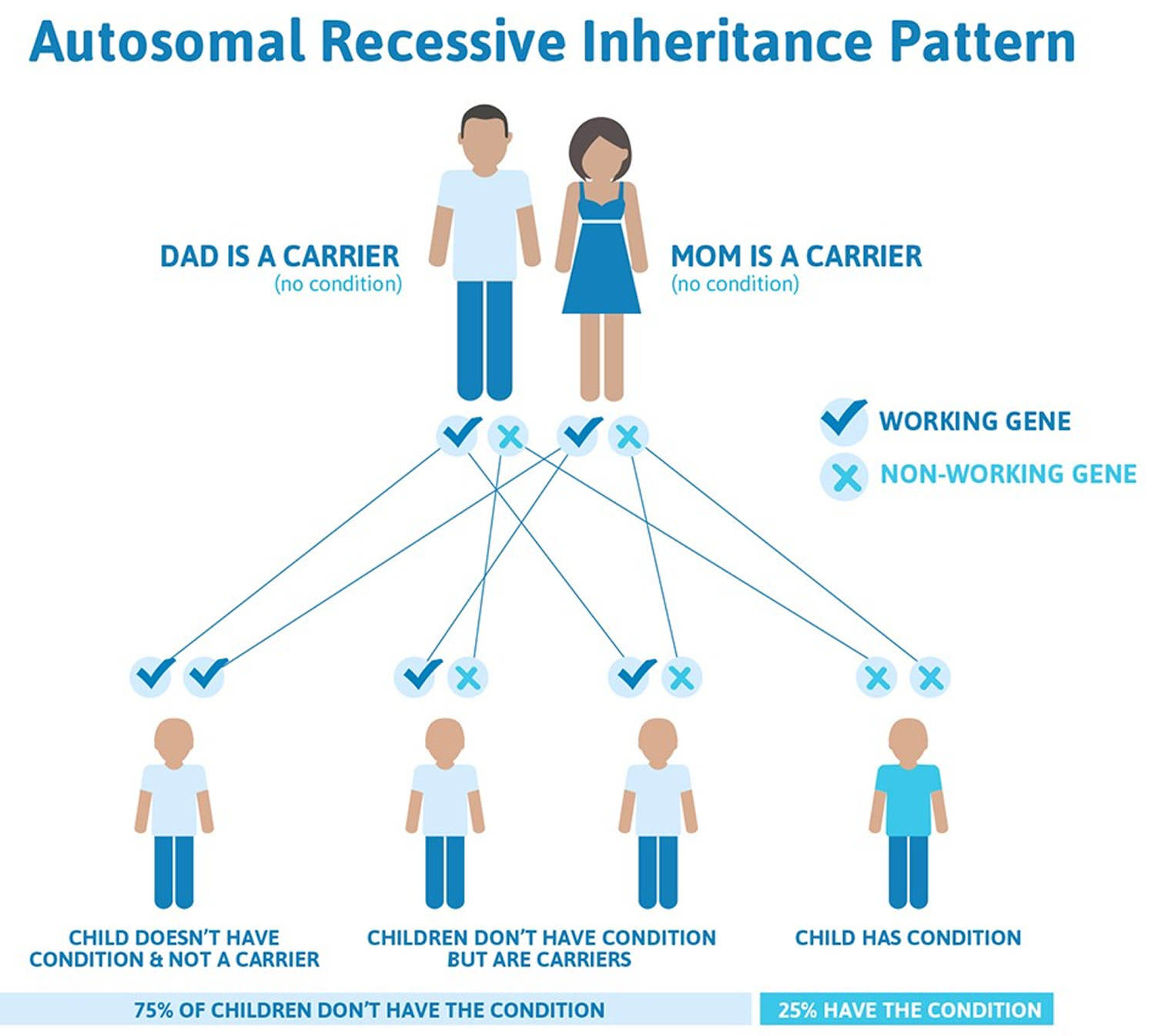
When due to genetic causes, this condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. Most often, the parents of an individual with an autosomal recessive disorder have one copy of the altered gene in each cell and are called carriers. They can pass on the gene mutation to their children, but they do not usually experience signs and symptoms of the disorder. In some cases, carriers of BCHE gene mutations take longer than usual to clear choline ester drugs from the body, but not as long as those with two copies of the altered gene in each cell.
Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Figure 1. Pseudocholinesterase deficiency carrier and autosomal recessive inheritance pattern
Acquired pseudocholinesterase deficiency causes
Neonates, elderly individuals, and pregnant women with certain physiologic conditions may have lower plasma pseudocholinesterase activity 12.
Pathologic conditions that may lower plasma pseudocholinesterase activity include the following 13:
- Chronic infections (tuberculosis)
- Extensive burn injuries
- Liver disease
- Kidney disease
- Heart attack/heart failure
- Malignancy 14
- Malnutrition
- Organophosphate pesticide poisoning
- Uremia
- Collagen diseases
- Myxedema (hypothyroidism)
One study recommended estimation of the pseudocholinesterase level to classify the severity of organophosphorous poisoning and to estimate prognosis. Pseudocholinesterase levels were reduced in all the cases in this study (N = 70), with the mean level being 3,154.16 ± 2,562.40 IU/L 15.
Pseudocholinesterase deficiency drugs to avoid
Iatrogenic causes of lower plasma pseudocholinesterase activity include plasmapheresis and medications such as the following:
- Anticholinesterase inhibitors
- Bambuterol
- Chlorpromazine
- Contraceptives
- Cyclophosphamide
- Echothiophate eye drops
- Esmolol
- Glucocorticoids
- Hexafluorenium
- Metoclopramide
- Monoamine oxidase inhibitors
- Pancuronium
- Phenelzine
- Tetrahydroaminacrine
Pseudocholinesterase deficiency diagnosis
Pseudocholinesterase deficiency test
Diagnosis of pseudocholinesterase deficiency is made by plasma assays of pseudocholinesterase enzyme activity. A sample of the patient’s plasma is incubated with the substrate butyrylthiocholine along with the indicator chemical 5,5′-dithiobis-(2-nitrobenzoic acid), which will produce a colored product that is assayed using spectrophotometry. The resulting amount of spectrophotometric absorption is proportionate to the pseudocholinesterase enzyme activity that is present in the patient’s plasma sample 16.
Because succinylcholine metabolites can interfere with this assay, plasma samples should be collected after muscle paralysis has completely resolved. The dibucaine and fluoride numbers can be determined by repeating this assay in the presence of standard aliquots of either dibucaine (0.03 mmol/L) or fluoride (4 mmol/L) in the reaction mixture to determine the percent inhibition of enzyme activity caused by these agents.
A simplified screening test of pseudocholinesterase enzyme activity can be performed using the Acholest Test Paper (see Table 2 below). When a drop of the patient’s plasma is applied to the substrate-impregnated test paper, a colorimetric reaction occurs. The time it takes the exposed Acholest Test Paper to turn from green to yellow is inversely proportional to the pseudocholinesterase enzyme activity in the plasma sample.
Table 2. Reaction Times for Acholest Test Paper
| Reaction Time | Pseudocholinesterase Enzyme Activity |
| < 5 min | Above normal |
| 5-20 min | Normal |
| 20-30 min | Borderline low |
| >30 min | Below normal |
The complete DNA sequence and amino acid structure of both the normal pseudocholinesterase protein and most of its abnormal variants have now been identified. Consultation with a geneticist may help to identify the specific atypical genotype alleles contributing to pseudocholinesterase deficiency.
Because the DNA sequence of the pseudocholinesterase gene and its amino acid structure is known, atypical alleles now can be identified by PCR amplification studies using DNA extracted from leukocytes in a blood sample. However, molecular genetic techniques such as polymerase chain reaction (PCR) amplification with allele-specific oligonucleotide probes for identifying abnormal pseudocholinesterase genotypes are currently available only in a limited number of research laboratories and are not in routine clinical use.
Pseudocholinesterase deficiency treatment
Pseudocholinesterase deficiency is a clinically silent condition in individuals who are not exposed to exogenous sources of choline esters.
While there is no cure for pseudocholinesterase deficiency, treatments are available. If breathing stops during surgery, breathing support can be provided. In most cases, recovery happens without the need for medical assistance.
Patients with prolonged paralysis following administration of succinylcholine can be treated in the following ways:
- Prophylactic transfusion of fresh frozen plasma can augment the patient’s endogenous plasma pseudocholinesterase activity. This practice is not recommended because of the risk of iatrogenic viral infectious complications. However, perioperative transfusion of fresh frozen plasma administered to correct a coagulopathy may mask an underlying pseudocholinesterase deficiency.
- Mechanical ventilatory support is the mainstay of treatment until respiratory muscle paralysis spontaneously resolves. Recovery eventually occurs as a result of passive diffusion of succinylcholine away from the neuromuscular junction.
- Administration of cholinesterase inhibitors, such as neostigmine, is controversial for reversing succinylcholine-related apnea in patients who are pseudocholinesterase deficient. The effects may be transient, possibly followed by intensified neuromuscular blockade.
If you have a family history of pseudocholinesterase deficiencyc or a family member who has problems with anesthesia, tell your doctor prior to any surgery. You can prevent problems by being tested before using the drug. If you have pseudocholinesterase deficiency, you may want to wear a medical bracelet identifying your condition. Tell family members to be tested prior to surgery as well.
- Pantuck E. Plasma cholinesterase: gene and variations. Anesth Analg. 1993;77:380–386. https://www.ncbi.nlm.nih.gov/pubmed/8346840[↩][↩]
- Zencirci B. Pseudocholinesterase enzyme deficiency: a case series and review of the literature. Cases Journal. 2009;2:9148. doi:10.1186/1757-1626-2-9148. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2803945/[↩]
- Daniel R Alexander. Pseudocholinesterase Deficiency. https://emedicine.medscape.com/article/247019-overview[↩]
- Lockridge O, Bartels CF, Vaughan TA, Wong CK, Norton SE, Johnson LJ. Complete amino acid sequence of human serum cholinesterase. J Biol Chem. 1987;262:549–557. http://www.jbc.org/content/262/2/549.long[↩]
- Pedersen NA, Jensen FS. Clinical importance of plasma cholinesterase for the anaesthetist. Ann Acad Med Singapore. 1994;23(Suppl):120–124. https://www.ncbi.nlm.nih.gov/pubmed/7710221[↩]
- Ostergaard D, Viby-Mogensen J, Hanel HK, Skovgaard LT. Halflife of plasma cholinesterase. Acta Anaesthesiol Scand. 1988;32:266–269. doi: 10.1111/j.1399-6576.1988.tb02727.x. https://www.ncbi.nlm.nih.gov/pubmed/3364151[↩]
- Cerf C, Mesguish M, Gabriel I, Amselem S, Duvaldestin P. Screening patients with prolonged neuromuscular blockade after succinylcholine and mivacurium. Anesth Analg. 2002;94:461–466. doi: 10.1097/00000539-200202000-00044. https://www.ncbi.nlm.nih.gov/pubmed/11812719[↩]
- Ali HH, Savarese JJ, Embree PB, Basta SJ, Stout RG, Bottros LH, Weakly JN. Clinical pharmacology of mivacurium chloride (BW B1090U) infusion: comparison with vecuronium and atracurium. Br J Anaesth. 1988;61:541–546. doi: 10.1093/bja/61.5.541. http://bjanaesthesia.org/article/S0007-0912(17)49230-0/pdf[↩]
- Savarese JJ, Lien CA, Belmont MR, Wastila WB. The clinical pharmacology of new benzylisoquinoline-diester compounds with special consideration of cisatracurium and mivacurium. Anesthetist. 1997;46:840–849. doi: 10.1007/s001010050477. https://www.ncbi.nlm.nih.gov/pubmed/9424966[↩]
- Goudsouzian NG, d’Hollander AA, Viby-Mogensen J. Prolonged neuromuscular block from mivacurium in two patients with cholinesterase deficiency. Anesth Analg. 1993;77:183–185. doi: 10.1213/00000539-199307000-00035. https://www.ncbi.nlm.nih.gov/pubmed/8317728[↩]
- Ellison M, Grose B, Howell S, Wilson C, Lenz J, Driver R. Prolonged Paralysis Following Emergent Cesarean Section with Succinylcholine Despite Normal Dibucaine Number. W V Med J. 2016 Mar-Apr. 112 (2):44-6.[↩]
- Brozović G, Mazul Sunko B, Hafner T, Bekavac I. Allergic reaction to suxamethonium during emergency caesarean section and pseudocholinesterase deficiency in the same patient. Wien Klin Wochenschr. 2014 Jul. 126 (13-14):435-8.[↩]
- Davis L, Britten JJ, Morgan M. Cholinesterase. Its significance in anaesthetic practice. Anaesthesia. 1997;52:244–260. doi: 10.1111/j.1365-2044.1997.084-az0080.x. https://www.ncbi.nlm.nih.gov/pubmed/9124666 https://onlinelibrary.wiley.com/doi/pdf/10.1111/j.1365-2044.1997.084-az0080.x[↩]
- LaRocca CJ, Beilman GJ, Birch M. A Case of Pseudocholinesterase Deficiency Resulting From Malnutrition. A A Case Rep. 2016 Sep 1. 7 (5):112-4.[↩]
- Chaudhary SC, Singh K, Sawlani KK, Jain N, Vaish AK, Atam V, et al. Prognostic significance of estimation of pseudocholinesterase activity and role of pralidoxime therapy in organophosphorous poisoning. Toxicol Int. 2013 Sep. 20(3):214-7.[↩]
- Duysen EG, Li B, Carlson M, Li YF, Wieseler S, Hinrichs SH, et al. Increased hepatotoxicity and cardiac fibrosis in cocaine-treated butyrylcholinesterase knockout mice. Basic Clin Pharmacol Toxicol. 2008 Dec. 103(6):514-21.[↩]
- Pseudocholinesterase Deficiency Workup. https://emedicine.medscape.com/article/247019-workup[↩]





{kind=link}