Contents
VACTERL association
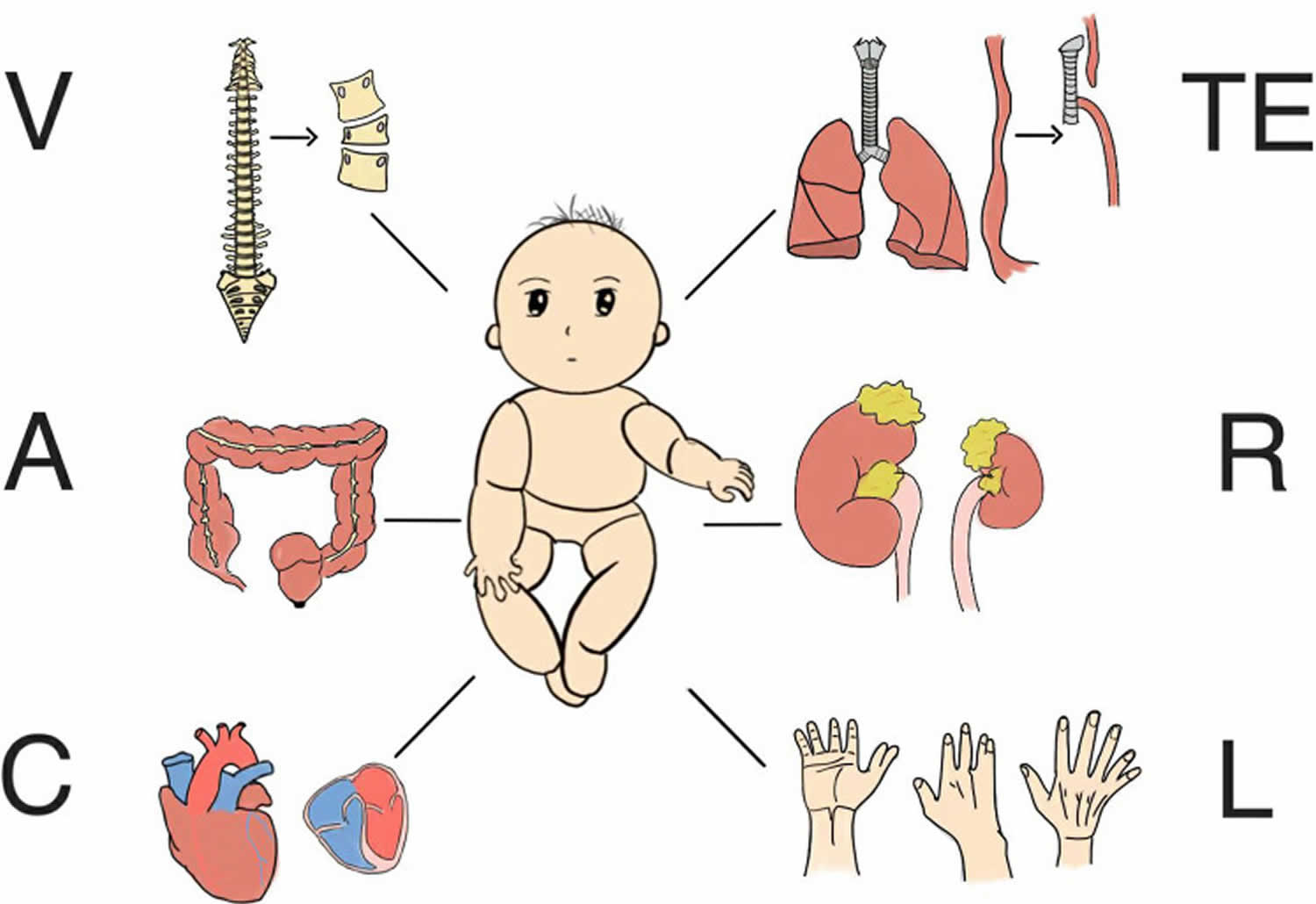
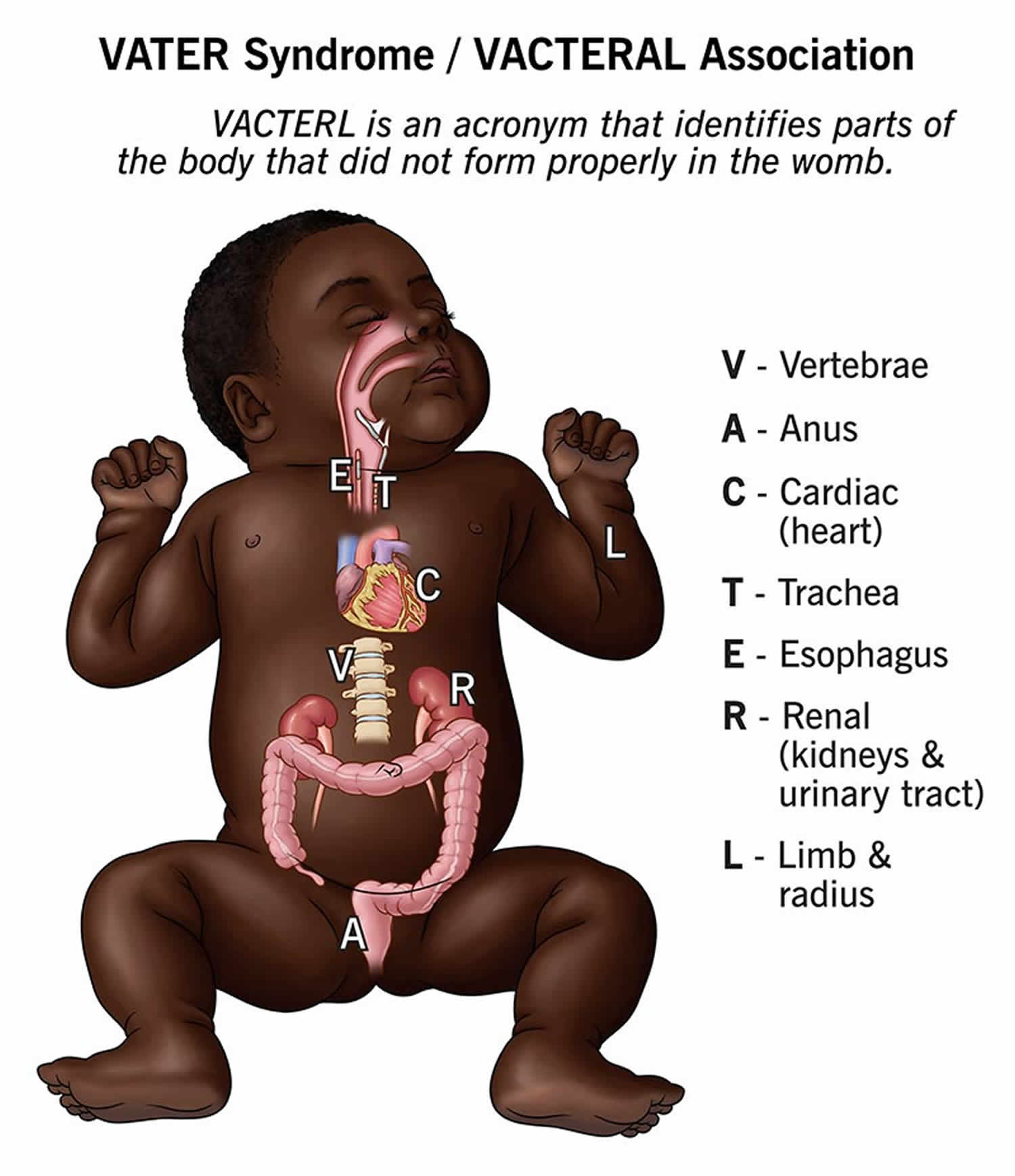
VACTERL association also known as VATER association is an abbreviation for rare group of congenital birth defects (birth defects that are present at birth) that affects multiple body parts that include 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17:
- V = Vertebral abnormalities with defects in the spine, such as misshapen or fused vertebrae or missing or extra vertebrae. Defects in the bones of the spine (vertebrae) are present in 60 to 80 percent of children with VACTERL association. Vertebral abnormalities may include misshapen vertebrae, fused vertebrae, and missing or extra vertebrae. In some people, spinal problems require surgery or cause other health problems, such as back pain of varying severity, throughout life.
- A = Anal atresia, a blockage or narrowing of the anus, which may also involve genitourinary anomalies. 55 to 90 percent of individuals with VACTERL association have narrowing or blockage of the anus (anal atresia). Anal atresia may be accompanied by abnormalities of the genitalia and urinary tract (genitourinary anomalies).
- C = Cardiac, heart defects, ranging from mild to severe. Heart (cardiac) defects occur in 40 to 80 percent of individuals with VACTERL association. Heart defects can range in severity from a life-threatening problem to a subtle defect that does not cause health problems e.g., atrial septal defect, ventricular septal defect, patent ductus arteriosus, hypoplastic left heart syndrome, transposition of the great arteries, and tetralogy of Fallot.
- TE = Trachea and Esophagus abnormalities, tracheo-esophageal fistula, a persistent connection between the windpipe and the esophagus. Fifty to 80 percent of people with VACTERL association have a tracheo-esophageal fistula, which is an abnormal connection (fistula) between the esophagus and the windpipe (trachea). Tracheoesophageal fistula can cause problems with breathing and feeding early in life and typically requires surgical correction in infancy. Clinical symptoms include episodic coughing after swallowing, progressively worsening dysphagia and dyspnea, difficulty inserting nasogastric tubes, and complications such as pneumonia and pleural effusion.
- R = Renal, kidney problems, which can include absent or malformed kidneys. Kidney (renal) anomalies occur in 50 to 80 percent of individuals with VACTERL association. Affected individuals may be missing one or both kidneys (renal aplasia) or have abnormally developed (renal hypoplasia) or misshapen kidneys (renal dysplasia), horseshoe kidney, cystic kidneys, and cystic dysplastic kidneys. Occasionally, ureteral and urogenital tract abnormalities may also be present, which can affect kidney function.
- L = Limb abnormalities, such as issues with the radius (radial agenesis) or other limb differences. Limb abnormalities are seen in 40 to 70 percent of people with VACTERL association. These abnormalities most commonly include poorly developed or missing thumbs or underdeveloped forearms and hands.
- S = Single umbilical artery in some cases.
VACTERL association diagnosis typically requires at least 3 of these these characteristic anatomical defects, though each person with VACTERL association is unique and can have different combination and severity of symptoms, often with additional abnormalities that are not among the characteristic features of VACTERL association. These defects are congenital (present at birth) and may be obvious at birth (e.g., anal atresia, tracheoesophageal fistula and esophageal atresia, radial defects) or not become recognized until later (e.g., cardiac, vertebral, and renal malformations).
Figure 1. VACTERL association
VACTERL association was first named in the early 1970’s by Quan and Smith 18, 19, 20. Because these characteristic birth defects were observed to occur together more often than would be expected by chance, the condition was termed an “association” 21. Furthermore, there was not and still remains no evidence for a single, unifying cause that would link all the birth defects together resulting in VACTERL being termed a syndrome 21. Furthermore, some authors have suggested that VACTERL association would be more accurately described as a “primary polytopic developmental field defect” as this reflects the causative developmental perturbation rather than an association as this simply describes the presence of statistical clustering 22, 23. One explanation for the clustering of features involves the idea of the “developmental field defect”, in which malformations that occur in blastogenesis tend to result in polytopic anomalies, or birth defects affecting multiple organ systems 21.
Several conditions have features in common with VACTERL association including 21:
- Alagille syndrome also known as Alagille-Watson syndrome, Watson-Miller syndrome, arteriohepatic dysplasia or syndromic bile duct paucity, is a congenital genetic multisystem disorder that affects the liver, heart, and other organs due to a shortage of bile ducts, which causes bile to build up and damage the liver 24. Alagille syndrome is caused by mutations in the JAG1 (90% of cases) or, less commonly, the NOTCH2 gene (1-2% of cases) and can lead to symptoms like jaundice, poor growth, and heart or kidney problems. Management focuses on treating symptoms and complications, with treatment options improving for those with early diagnosis
- Baller-Gerold syndrome is a rare genetic disorder characterized by premature fusion of the skull bones (craniosynostosis) and malformations of the forearm and hand bones 25. Other symptoms can include slow growth, a characteristic skin rash (poikiloderma), and a risk of developing certain cancers 26, 25. Skin lesions may appear anytime within the first few years after birth, typically beginning with erythema of the face and extremities and evolving into poikiloderma. Slow growth is apparent in infancy with eventual height and length typically at 4 standard deviation below the mean. Baller-Gerold syndrome is an autosomal recessive condition, meaning both parents must pass on a mutated gene, most commonly in the RECQL4 gene, for the child to be affected.
- CHARGE syndrome is a rare genetic condition affecting multiple body systems, characterized by a combination of major features: Coloboma (eye tissue missing), Heart defects, Atresia of the choanae (blocked nasal passages), Retardation of growth and development, Genital abnormalities, and Ear anomalies, including hearing loss 27, 28, 29. According to updated diagnostic criteria for CHARGE syndrome, the most defining features are the 4 Cs: Coloboma (eye tissue missing); Choanal atresia (blocked nasal passages); Cranial nerve anomalies (especially olfactory pathway absence) and characteristic ear anomalies (especially semicircular canal dysplasia/aplasia). CHARGE syndrome is caused by a genetic change, most often in the CHD7 gene, and while there is no cure, treatments can help manage symptoms.
- Currarino syndrome also known as the Currarino triad or ASP triad is a rare, autosomal dominant inherited condition characterized by a combination of three key features: an anorectal malformation, a sacrococcygeal (tailbone) defect, and a presacral mass (a lump in front of the sacrum) 30, 31, 32, 33. Currarino syndrome is usually caused by a mutation in the MNX1 gene, and often presents in childhood with symptoms like chronic constipation. Treatment depends on the specific anomalies present and can range from conservative management to surgery.
- Fanconi anemia is a rare, inherited genetic disorder that leads to progressive bone marrow failure, causing symptoms like fatigue, bleeding, and frequent infections 34, 35. In people with Fanconi anemia, impaired bone marrow function leads to a decrease in the production of all blood cells (aplastic anemia). Affected individuals experience extreme tiredness (fatigue) due to low numbers of red blood cells (anemia), frequent infections due to low numbers of white blood cells (neutropenia), and clotting problems due to low numbers of platelets (thrombocytopenia). People with Fanconi anemia may also develop myelodysplastic syndrome, a condition in which immature blood cells fail to develop normally. Individuals with Fanconi anemia have an increased risk of developing a cancer of blood-forming cells called acute myeloid leukemia (AML). They are also at risk of developing tumors of the liver, gastrointestinal system, genital tract, or head and neck known as head and neck squamous cell carcinoma. The likelihood of a person with Fanconi anemia developing one of these cancers is between 10 and 30 percent 34. People with Fanconi anemia often have growth problems before or after birth that often result in short stature. Affected individuals can also have irregular skin coloring such as unusually light-colored skin (hypopigmentation) or café-au-lait spots, which are flat patches on the skin that are darker than the surrounding area. People with Fanconi anemia can have skeletal problems that often include malformed thumbs or forearms or an unusually small head size (microcephaly). Problems in hormone-producing (endocrine) tissues are common in people with Fanconi anemia, including abnormally low levels of thyroid hormones (hypothyroidism), and high blood glucose levels (hyperglycemia). Individuals with Fanconi anemia can also have eye abnormalities such as small or abnormally shaped eyes or malformed or absent kidneys and other defects of the urinary tract. Less frequent problems include gastrointestinal abnormalities, heart defects, brain abnormalities, and hearing loss. People with Fanconi anemia may have abnormal genitalia or malformations of the reproductive system, which can result in difficulty having biological children (infertility).
- Holt-Oram syndrome also called “hand-heart syndrome” is a autosomal dominant genetic disorder caused by a mutation in the TBX5 gene that affects hands and arms (upper limbs) development and causes congenital heart defects 36, 37, 38. Common symptoms include abnormalities of the bones in the arms and hands, such as a missing or malformed thumb, and heart problems like atrial or ventricular septal defects and conduction abnormalities. People with Holt-Oram syndrome have abnormally developed bones in their upper limbs. At least one abnormality in the bones of the wrist (carpal bones) is present in affected individuals. Often, these wrist bone abnormalities can be detected only by x-ray. Individuals with Holt-Oram syndrome may have additional bone abnormalities including a missing thumb, a long thumb that looks like a finger, partial or complete absence of bones in the forearm, an underdeveloped bone of the upper arm, and abnormalities of the collar bone or shoulder blades. These skeletal abnormalities may affect one or both of the upper limbs. If both upper limbs are affected, the bone abnormalities can be the same or different on each side. In cases where the skeletal abnormalities are not the same on both sides of the body, the left side is usually more severely affected than the right side. About 75 percent of individuals with Holt-Oram syndrome have heart (cardiac) problems, which can be life-threatening. The most common problem is a defect in the muscular wall (septum) that separates the right and left sides of the heart. A hole in the septum between the upper chambers of the heart (atria) is called an atrial septal defect (ASD), and a hole in the septum between the lower chambers of the heart (ventricles) is called a ventricular septal defect (VSD). Some people with Holt-Oram syndrome have cardiac conduction disease, which is caused by abnormalities in the electrical system that coordinates contractions of the heart chambers. Cardiac conduction disease can lead to problems such as a slower-than-normal heart rate (bradycardia) or a rapid and uncoordinated contraction of the heart muscle (fibrillation). Cardiac conduction disease can occur along with other heart defects (such as ASD or VSD) or as the only heart problem in people with Holt-Oram syndrome. The features of Holt-Oram syndrome are similar to those of a condition called Duane-radial ray syndrome; however, these two disorders are caused by mutations in different genes.
Furthermore, several conditions have been associated with VACTERL, including gray platelet syndrome in neonates 39, Omenn syndrome 40 and spinal muscular atrophy 41.
The cause of VACTERL association remains unknown and is likely caused by a combination of different factors (multifactorial) and it is not considered a hereditary disorder, which means they occur in people with no history of the condition in their family 42, 43. A study of 25 newborns with VACTERL association in Mexico reported that the risk factors for VACTERL association were gestational diabetes and consumption of statins, cocaine, or marijuana 44. VACTERL association is generally not a heritable disease, and the risk of recurrence in another child from the same parents is low.
Most cases of VACTERL association are sporadic, which means they occur in people with no history of the condition in their family. Rarely, families have multiple people affected with VACTERL association. A few affected individuals have family members with one or two features, but not enough signs to be diagnosed with the condition. In these families, the features of VACTERL association often do not have a clear pattern of inheritance. Multiple genetic and environmental factors likely play a part in determining the risk of developing this condition and how severe the condition will be in an individual.
VACTERL association is estimated to occur in about 1 in 10,000 to 1 in 40,000 live-born infants 21. According to data published by the European Commission, the prevalence of VACTERL association was 0.47 to 0.58 per 10,000 live births between 2012 and 2022 45. However, the true frequency of VACTERL association may be difficult to determine because different diagnostic criteria are used in different studies. In addition, VACTERL association is likely to be underdiagnosed, especially in children with fewer problems. Some studies have shown that males might be slightly more commonly affected than females, but no association with a specific geographic region or ethnic group has been determined 21.
There is no one test for VACTERL association, also known as VATER syndrome. Instead, it’s considered a “diagnosis of exclusion”. That means that when other causes of the birth defects have been ruled out, the remaining diagnosis is VACTERL association. To rule out other causes with similar symptoms, doctors will visually assess the patient and run a series of tests, including genetics.
Some of the features of VACTERL association can be subtle and are not identified until late in childhood or adulthood, making diagnosis of this condition difficult.
The treatment of VACTERL association is directed toward the specific birth defects and related symptoms that occur in each individual, which often varies greatly. Many of the structural abnormalities (kidney defects, heart defects, anal atresia, etc.) can be surgically corrected. In some individuals, surgery might be necessary as early as the neonatal period. Repeat surgeries might also be needed later in life to revise or further correct structural defects.
Because VACTERL association is a complex condition, its management and care include a coordinated care from a team of specialists. Individuals diagnosed with VACTERL association will need to be followed by a number of medical and developmental specialists depending on their individual needs. Some of the medical specialists who often follow individuals with VACTERL association include cardiologists, urologists, orthopedists, ear, nose and throat (ENT) physicians and clinical geneticists. Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Is VACTERL association a hereditary condition?
No. VACTERL association isn’t a hereditary condition because the condition usually only affects one person in a family (sporadic). Multiple genetic and environmental factors determine who is at risk of acquiring VACTERL association and the severity of their symptoms.
How do I take care of my child diagnosed with VACTERL association?
Your child with VACTERL association will need ongoing treatment throughout their life to alleviate symptoms of the condition. As their caregiver, make sure you keep track of your child’s health and growth during childhood to make sure they don’t miss developmental milestones. Stay up to date with your child’s doctors, which could include a team of specialists to treat your child’s unique symptoms.
Some families find support through genetic counseling to help them navigate their child’s diagnosis and care options.
VACTERL association causes
VACTERL association is a complex condition that may have different causes in different people 42, 43. In some people, VACTERL association is likely caused by the interaction of multiple genetic and environmental factors. Some possible genetic and environmental influences have been identified and are being studied. For example, a study of 25 newborns with VACTERL association in Mexico reported that the risk factors for VACTERL association were gestational diabetes and consumption of statins, cocaine, or marijuana 44. The developmental abnormalities characteristic of VACTERL association develop before birth. The disruption to fetal development that causes VACTERL association likely occurs early in development, resulting in birth defects that affect multiple body systems. It is unclear why the features characteristic of VACTERL association group together in affected individuals.
Most cases of VACTERL association are sporadic, which means they occur in people with no history of the condition in their family. Rarely, families have multiple people affected with VACTERL association. A few affected individuals have family members with one or two features, but not enough signs to be diagnosed with VACTERL association. In these families, the features of VACTERL association often do not have a clear pattern of inheritance. Multiple genetic and environmental factors likely play a part in determining the risk of developing VACTERL association and how severe the condition will be in an individual.
Rarely, VACTERL association has been associated with gene alterations including duplications or deletions (copy number variation), and mitochondrial dysfunction (the mitochondria is a cellular structure responsible for energy production in the cell).
At the molecular level, current research focuses on Sonic Hedgehog (SHH) signaling pathways, cilia-associated signaling pathways, and other genes influencing embryonic development. Maternal gestational status, the fetal environment, and the use of assisted reproductive techniques (ARTs) may also contribute to VACTERL development 1, 46, 47, 48, 49. These factors influence fetal growth and organogenesis, ultimately contributing to the development of VACTERL 1.
VACTERL association signs and symptoms
VACTERL association also known as VATER association is an abbreviation for rare group of congenital birth defects (birth defects that are present at birth) that affects multiple body parts that include 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 50, 51, 52, 15:
- V = Vertebral abnormalities with defects in the spine, such as misshapen or fused vertebrae or missing or extra vertebrae. Defects in the bones of the spine (vertebrae) are present in 60 to 80 percent of people with VACTERL association. Vertebral abnormalities may include misshapen vertebrae, fused vertebrae, and missing or extra vertebrae. In some people, spinal problems require surgery or cause other health problems, such as back pain of varying severity, throughout life.
- A = Anal atresia, a blockage or narrowing of the anus, which may also involve genitourinary anomalies. 60 to 90 percent of individuals with VACTERL association have narrowing or blockage of the anus (anal atresia). Anal atresia may be accompanied by abnormalities of the genitalia and urinary tract (genitourinary anomalies).
- C = Cardiac, heart defects, ranging from mild to severe.
- TE = Trachea and Esophagus abnormalities, tracheo-esophageal fistula, a persistent connection between the windpipe and the esophagus.
- R = Renal, kidney problems, which can include absent or malformed kidneys.
- L = Limb abnormalities, such as issues with the radius (radial agenesis) or other limb differences.
- S = Single umbilical artery in some cases.
VACTERL association diagnosis typically requires at least 3 of these these characteristic anatomical defects, though each person with VACTERL association is unique and can have different combination and severity of symptoms, often with additional abnormalities that are not among the characteristic features of VACTERL association. These defects are congenital (present at birth) and may be obvious at birth (e.g., anal atresia, tracheoesophageal fistula and esophageal atresia, radial defects) or not become recognized until later (e.g., cardiac, vertebral, and renal malformations).
VACTERL association involves multiple anatomical defects that may vary greatly from one child to another and an affected child will typically not have all of the malformations listed below.
Vertebral anomalies
As many as 80% of people with VACTERL association have defects or anomalies in the bones of their spine, sometimes also involving the ribs. These defects can include missing vertebrae, malformed vertebrae (half-formed vertebrae termed hemivertebrae, butterfly-shaped vertebrae, vertebral clefts and fusion of vertebrae), missing ribs, an increased number of ribs (supernumerary ribs), rib fusions and splitting of ribs. Side-to-side curvature of the spine (scoliosis) and absence of the tailbone, the lowest bone of the spinal column (sacral agenesis), may also occur.
Anal Atresia
The majority of people with VACTERL association have an abnormal anus, including anal atresia also called imperforate anus, where the opening that connects the rectum through the anus to the exterior is closed, missing or blocked. The anal closure may be a thin membrane of skin or a thicker blockage of skin and muscle. Anal atresia or imperforate anus prevents the normal passage of bowel contents. Anal atresia may coexist with abnormalities in the lower urogenital tract. Signs of anal atresia include a bloated abdomen, vomiting, and a lack of or light and irregular bowel movements.
Cardiac (heart) defects
Roughly half of the children with VACTERL association will have some abnormalities of the heart. These can take many forms, but the most common are 53:
- Atrial septal defect (ASD) — a hole in the wall between the two upper chambers of the heart
- Hypoplastic left heart syndrome (HLHS) —a malformation of the left side of the heart which prevents blood from flowing normally through the heart
- Patent ductus arteriosus (PDA) — an abnormal opening in a blood vessel in the heart keeps blood from traveling to the lungs for oxygenation
- Tetralogy of Fallot — a combination of four heart defects that affect the aortic valve, pulmonary valve and right ventricle
- Transposition of the great arteries —two of the heart’s main arteries are backward
- Ventricular septal defect (VSD) — a hole in the wall that divides the right and left lower chambers of the heart (ventricles). Ventricular septal defect (VSD) is the most common heart defect in VACTERL association.
Normal heart has four chambers. The two upper chambers, known as atria, are separated from each other by a fibrous partition known as the atrial septum. The two lower chambers are known as ventricles and are separated from each other by the ventricular septum. Valves connect the atria (left and right) to their respective ventricles. The aorta, the main vessel of arterial circulation, carries blood away from the left ventricle to the rest of the body. A ventricular septal defect (VSD) is a hole in the ventricular septum and may occur anywhere in the septum. The size and location of the defect determine the severity of the symptoms. A small ventricular septal defect may close on its own (spontaneously) or become less significant as the child matures and grows. A moderately-sized defect may affect the ability of the heart to pump blood efficiently to the lungs and the rest of the body (congestive heart failure). Symptoms associated with heart failure include an abnormally rapid rate of breathing (tachypnea), wheezing, an unusually fast heartbeat (tachycardia), and failure to grow at the expected rate (failure to thrive). A large ventricular septal defect may cause life-threatening complications during infancy.
Additional congenital heart defects that have occurred in the VACTERL association include atrial septal defects (ASDs); hypoplastic left heart syndrome (a life-treating condition in which there is underdevelopment of the left ventricle, the aortic and/or mitral valve, and the ascending aorta); transposition of the great arteries (a condition in which the aorta and pulmonary artery are switched in positions); a complex malformation known as tetralogy of Fallot; and patent ductus arteriosus [a condition in which the passage between the blood vessel that leads to the lungs (pulmonary arteries) and the major artery of the body (aorta) fails to close after birth].
Signs that a child has heart anomalies can include shortness of breath or difficulty breathing, fatigue, blue skin color, an abnormal heart rhythm, heart murmur, fast pulse and failure to gain weight/poor appetite.
Trachea and esophageal anomalies
The trachea is more commonly called your windpipe; the esophagus also called foodpipe is the tube that food enters when you swallow. Normally, these tubes are not connected in any way, but for children with VACTERL association, an abnormal connection between the trachea and the esophagus called tracheoesophageal fistula (a hole or passage between the trachea and the esophagus). The tracheoesophageal fistula potentially allows food to be inhaled (aspirated) into the lungs, which, in turn, may result in respiratory infections (e.g., pneumonia) and failure to thrive.
Children with VACTERL association may have other abnormalities, such as esophageal atresia which is a malformation in which the esophagus narrows to a thin cord or ends in a blind pouch and doesn’t properly connect to the stomach. The tracheoesophageal fistula and esophageal atresia may result in breathing, feeding and swallowing difficulties including coughing or choking while eating, aspirating food into the lungs, blue color, poor appetite and weight gain, swollen abdomen and vomiting.
Renal (kidney) anomalies
About half of all individuals with VACTERL association have issues with their kidneys and urinary tract. These can take many forms including absence of development of one or both kidneys (renal aplasia), malformation of one or both kidneys (renal dysplasia), displaced or malpositioned kidneys (renal ectopia), abnormal backflow (reflux) of urine into the tube (ureter) that carries urine to the bladder (vesicoureteral reflux), resulting in abnormal accumulation of urine in the kidneys (hydronephrosis). Children with any of these symptoms are more prone to urinary tract infections. For boys, they may also have hypospadias, where the urethral opening is on the bottom of the penis instead of the tip.
Limb Anomalies
Another major finding associated with VACTERL association is defects affecting the forearm on the thumb side (radius). These defects may include absence of the radius (radial aplasia), underdevelopment of the radius (radial hypoplasia), underdevelopment or absence of the thumb and/or the presence of an extra bone in the thumb (triphalangeal thumb). Other limb anomalies including extra digits (polydactyly), webbing of the digits (syndactyly), abnormal fusion of the two forearm bones (radioulnar synostosis) and lower limb malformations such as clubfoot, and hypoplasia of the great toe and tibia have been described in VACTERL association.
Other Birth Defects
Additional birth defects that have been reported to occur in a minority of VACTERL association individuals include facial asymmetry (hemifacial microsomia), abnormal shape and size of the ears, narrowing of the larynx (laryngeal stenosis), narrowing of the passages from the back of the nose to the throat that make it possible to breathe through the nose (choanal atresia), lung malformations, protrusion of part of the intestines through an abnormal opening in the muscular abdominal wall near the umbilical cord (omphalocele), diaphragmatic hernia, intestinal misplacement and misalignment (malrotation), widening of the posterior fossa, hydrocephalus, cerebellar malformations, cerebral hypoplasia, cervical lymphangioma, pulmonary cystadenoma or sequestration, pulmonary hypoplasia, and tethered spinal cord. Other associated conditions include absent or hypoplastic nasal bones, cleft lip and/or palate, hearing loss, abnormal arteries, moyamoya disease, congenital intestinal malrotation, duodenal stenosis or atresia, single umbilical artery instead of the usual two arteries, umbilical cord cyst, umbilical hernia, persistent right umbilical vein, congenital genital abnormalities, cryptorchidism, ambiguous genitalia 54, intrauterine growth restriction 55, pancreatic structural anomalies 56, and biliary tract abnormalities 57.
Some infants and young children with VACTERL association may grow slower than normal. In the vast majority of patients, VACTERL association does not affect mental functioning and intelligence.
VACTERL association diagnosis
VACTERL association diagnosis is a clinical diagnosis based on the defects present and therefore starts with a complete physical examination.
- V = Vertebral abnormalities with defects in the spine, such as misshapen or fused vertebrae or missing or extra vertebrae. Defects in the bones of the spine (vertebrae) are present in 60 to 80 percent of people with VACTERL association. Vertebral abnormalities may include misshapen vertebrae, fused vertebrae, and missing or extra vertebrae. In some people, spinal problems require surgery or cause other health problems, such as back pain of varying severity, throughout life.
- A = Anal atresia, a blockage or narrowing of the anus, which may also involve genitourinary anomalies. 60 to 90 percent of individuals with VACTERL association have narrowing or blockage of the anus (anal atresia). Anal atresia may be accompanied by abnormalities of the genitalia and urinary tract (genitourinary anomalies).
- C = Cardiac, heart defects, ranging from mild to severe.
- TE = Trachea and Esophagus abnormalities, tracheo-esophageal fistula, a persistent connection between the windpipe and the esophagus.
- R = Renal, kidney problems, which can include absent or malformed kidneys.
- L = Limb abnormalities, such as issues with the radius (radial agenesis) or other limb differences.
- S = Single umbilical artery in some cases.
The diagnosis of VACTERL association is made when other possible causes of birth defects have been ruled out (diagnosis of exclusion).
Currently, there is no validated diagnostic criteria or universally accepted diagnostic standard for the VACTERL association and no laboratory test exists that can diagnose or rule out VACTERL association 1. Most clinicians and researchers agree that a diagnosis is warranted if a baby exhibits at least three of the six characteristic congenital abnormalities: vertebral defects, anal atresia, cardiac defects, tracheoesophageal malformation, renal defects, and limb anomalies 50. However, that criteria has not reached full acceptance, with some researchers and clinicians believing that 2 major malformations and one associated feature are sufficient 58. At the moment, the most stringent approach defines a “secure” designation of VACTERL association in presence of at least one anomaly in all three involved body parts (i.e. limbs, thorax and pelvis/lower abdomen), and “probable” in presence of two or more anomalies in two body parts.
Some researchers have mentioned that the presence of more than three malformations is not necessarily VACTERL association and that other neonatal malformations may be present if they are more pronounced and genetically compatible, e.g., a child with concurrent heart defects, anal atresia, vertebral anomalies with early-onset epilepsy, global developmental delay with autistic features, cerebellar hypoplasia, and characteristically dysmorphic facial features (slanted head with downward sloping blepharophimosis, short neck with webbing), the presence of heterozygous de novo missense variants of the PACS2 gene should be considered to be on the PACS2 spectrum of disorders 59. An incomplete expression of VACTERL is termed partial VACTERL (pVACTERL) 60.
Other tests performed will depend on the suspected birth defects. For example, X-ray imaging might be used to detect spine and limb anomalies and ultrasound imaging might identify heart or kidney defects. Esophageal atresia and tracheoesophageal fistula can also be detected on x-ray, and radiography is the preferred method for confirming tracheoesophageal malformations 50. Laboratory and genetic tests can be useful to rule out alternative diagnoses. Some malformations seen in VACTERL association might be identified before birth with imaging techniques such as prenatal ultrasound. Importantly, the presence of a single umbilical artery should prompt evaluation for VACTERL association and other birth defects.
VACTERL association differential diagnosis
The malformations that occur in VACTERL association can be seen in a number of other disorders. To avoid misdiagnosis, a complete evaluation is required to consider other possible causes of congenital malformations. Some of the disorders that can mimic VACTERL association are heritable and some occur sporadically. Examples of such conditions are described below.
VACTERL with hydrocephalus is an extremely rare genetic disorder in which the multisystem features of VACTERL association occur in addition to hydrocephalus. Hydrocephalus is a condition in which excessive accumulation of cerebrospinal fluid in the skull causes pressure on the tissues of the brain and may result in abnormally enlarged head size (macrocephaly). Additional common symptoms associated with hydrocephalus include vomiting, irritability, seizures, and downward gaze of the eyes (sunsetting). Some affected infants may experience delays in reaching developmental milestones (developmental delays). The specific birth defects that occur in VACTERL with hydrocephalus vary from one child to another. Unlike VACTERL association, VACTERL with hydrocephalus is a genetic disorder and may be inherited in an autosomal recessive or X-linked pattern.
Baller-Gerold syndrome is a rare genetic disorder characterized by premature fusion of the skull bones (craniosynostosis) and malformations of the forearm and hand bones 25. Other symptoms can include slow growth, a characteristic skin rash (poikiloderma), and a risk of developing certain cancers 26, 25. Skin lesions may appear anytime within the first few years after birth, typically beginning with erythema of the face and extremities and evolving into poikiloderma. Slow growth is apparent in infancy with eventual height and length typically at 4 standard deviation below the mean. Baller-Gerold syndrome is an autosomal recessive condition, meaning both parents must pass on a mutated gene, most commonly in the RECQL4 gene, for the child to be affected.
Fanconi anemia is an inherited anemia that leads to progressive, severe bone marrow failure, also known as aplastic anemia. The disorder is characterized by weakness, severe bleeding due to insufficient blood clotting and susceptibility to infection. Fanconi anemia may also be associated with heart (cardiac), kidney (renal), and/or skeletal abnormalities. Patients are also at increased risk for developing leukemia and other cancers. There are several different subtypes (complementation groups) of Fanconi anemia, each of which is thought to result from an abnormal change (mutation) to a different gene. Fanconi anemia follows autosomal recessive inheritance.
CHARGE syndrome is a rare pattern of malformations that may affect several organ systems of the body. CHARGE is an acronym that stands for Coloboma of the eye; Heart defects; Atresia of the Choanae, meaning bony or membranous blockage of the passageway between the nose and throat; Retardation of growth and development and/or mental deficiency; Genital anomalies; and Ear anomalies and/or deafness. Some affected individuals may also have other findings such as a small head (microcephaly), incomplete closure of the roof of the mouth (cleft palate), an abnormal groove in the upper lip (cleft lip), swallowing difficulties, paralysis of facial nerves (facial palsy), an abnormal connection between the windpipe and the tube that carries food from the throat to the stomach (tracheoesophageal fistula) and renal malformations. CHARGE syndrome is caused by alterations in the CHD7 gene.
Currarino syndrome also known as the Currarino triad or ASP triad is a rare, autosomal dominant inherited condition characterized by a combination of three key features: an anorectal malformation, a sacrococcygeal (tailbone) defect, and a presacral mass (a lump in front of the sacrum) 30, 31, 32, 33. Currarino syndrome is usually caused by a mutation in the MNX1 gene, and often presents in childhood with symptoms like chronic constipation. Treatment depends on the specific anomalies present and can range from conservative management to surgery.
Holt-Oram syndrome is a rare genetic disorder characterized by distinctive malformations of the bones of the thumbs and forearms and the heart. Holt-Oram syndrome is an autosomal dominant disorder and as such may be inherited from an affected parent. In about 40 percent of patients, the disorder is the result of a spontaneous (i.e., de novo) genetic change (i.e., new gene alteration). The disorder is associated with alterations in the TBX5 gene.
Townes-Brocks syndrome is a rare inherited disorder that is apparent at birth (congenital). Although symptoms and physical characteristics associated with Townes-Brocks syndrome may vary greatly in range and severity from person to person, abnormalities tend to involve the face, ears, arms and legs (limbs), gastrointestinal system, and kidneys 61, 62. Some affected individuals may also have abnormalities of the heart and the reproductive organs. Townes-Brocks syndrome follows autosomal dominant pattern of inheritance and in most patients is associated with alterations in the gene SALL1.
Trisomy 18 (Edwards syndrome) is a condition where affected individual have a third copy of chromosome 18 in their cells, compared to the normal two. It shows a significant overlap with VACTERL association, especially in the prenatal period and at birth. In fact, the combination of upper limb anomalies, congenital heart defects, renal malformations and growth retardation of prenatal onset is quite common in both conditions. Marked developmental delay easily differentiates the conditions later in infancy.
VACTERL association treatment
The treatment of VACTERL association is directed toward the specific birth defects and related symptoms that occur in each individual, which often varies greatly. Many of the structural abnormalities (kidney defects, heart defects, anal atresia, etc.) can be surgically corrected. In some individuals, surgery might be necessary as early as the neonatal period. Repeat surgeries might also be needed later in life to revise or further correct structural defects.
A team approach is essential for optimal treatment of the condition. Individuals diagnosed with VACTERL association will need to be followed by a number of medical and developmental specialists depending on their individual needs. Some of the medical specialists who often follow individuals with VACTERL association include cardiologists, urologists, orthopedists, ear, nose and throat (ENT) physicians and clinical geneticists.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
Anesthesia in children with VACTERL association also requires special attention because patients with VACTERL association are at elevated risk for anesthesia, such as tracheoesophageal fistula which can complicate airway management and preoperative aspiration, heart malformations that can affect hemodynamic stability, kidney anomalies that may cause abnormalities in pharmacokinetics and pharmacodynamics, and vertebral malformations that may cause difficulty in surgical positioning. The technique used during anesthesia may be an ultrasound-guided caudal block, which has been shown in some studies to improve the probability of a successful first puncture, and real-time ultrasound monitoring of local anesthetic spread also permits visual confirmation of correct placement. The use of ketamine and dexmedetomidine for sedation and analgesia has proven to be beneficial as it allows for balanced and titratable levels of sedation while maintaining voluntary ventilation. It also provides effective sedation and hemodynamic stability. The risk of respiratory depression and airway complications is minimized by avoiding volatile drugs and opioids 63. Post-surgical care should focus on airway management, gastric tube support, feeding and nutrition management, oral rehabilitation exercises, and maintaining airway patency and assisted ventilation, with emphasis on deoxygenation training.
VACTERL association prognosis
With the improvement of medical and surgical care, the prognosis or patient outcomes of VACTERL association are much better than they were before. However, affected individuals may experience medical complications throughout life 64, 65. For instance, vertebral malformations might lead to scoliosis and chronic back pain, anal atresia might be associated with incontinence and/or constipation, gastro-esophageal reflux might result from tracheoesophageal fistula and kidney anomalies are associated with an increased risk of urinary tract infection (UTI) and kidney stones (nephrolithiasis) 64, 66. Patients who have had tracheoesophageal fistula may be frequently hospitalized in childhood due to lung infections or stuck food. They take more time than their peers to complete a meal (affecting work-school life) and always have to consider the type of food they eat 67. Sixty percent of parents of children with tracheoesophageal fistula exhibit fear of choking 67. Also, frequent hospital admissions can have negative effects such as anxiety, and quality of life 68. Sometimes some patients do not develop other symptoms associated with VACTERL until adulthood. The inconvenience of daily life and physical pain may seriously affect patients’ quality of life and mental health. In addition, individuals with limb abnormalities and malformation might have functional limitations.
VACTERL association life expectancy
Fortunately, the symptoms associated with VACTERL association generally aren’t life-threatening and can be successfully treated, although some children may need multiple surgeries to achieve full function. Depending on their specific symptoms, children with ACTERL association may need ongoing supportive care and to be monitored by one or more specialties, including gastroenterology, urology, cardiology and orthopedics.
The long-term outlook for children with VACTERL association who have received treatment as needed for their specific symptoms is generally good and most experience typical intellectual development. However, some children with VACTERL association may be more prone to chronic health problems into adulthood. The good news is that with the right supportive care, patients with VACTERL association can lead happy, productive lives. Complication rates and mortality in VACTERL association patients depend on a variety of factors related to the patient’s condition, associated anomalies, surgical technique, and other factors.
- Sun M, Zhao Q, Yang B, Liu L, Zhou C, Yao X, Bu J, Bian J, Ge S, Zhu Z, Liu B. Molecular mechanism, diagnosis, and treatment of VACTERL association. Front Pediatr. 2025 Jul 7;13:1609624. doi: 10.3389/fped.2025.1609624[↩][↩][↩][↩][↩]
- Ritter J, Lisec K, Klinner M, Heinrich M, von Schweinitz D, Kappler R, Hubertus J. Genetic Disruption of Cilia-Associated Signaling Pathways in Patients with VACTERL Association. Children (Basel). 2023 May 14;10(5):882. doi: 10.3390/children10050882[↩][↩]
- Delgado J, Atkins L, Pippin M, Jishu J. A Case of a Newborn Presenting With a VACTERL-Like Association. Cureus. 2024 Dec 9;16(12):e75400. doi: 10.7759/cureus.75400[↩][↩]
- Oz-Alcalay L, Klinger G, Sokolover N, Merlob P, Scheinfeld T. Esophageal Lung and VACTERL Association Combined with Dysmorphic Features. Isr Med Assoc J. 2024 Dec;26(11):701-703. https://ima-files.s3.amazonaws.com/655652_b0f391b9-73b7-4370-a464-f23b844bde21.pdf[↩][↩]
- Soyer T, Pederiva F, Dalena P, Pio L, Kakar M, Hall NJ, Morini F; Esophageal Atresia Registry participants. Impact of VACTERL Association and Chromosomal Anomalies on Outcomes After Esophageal Atresia Repair: Insights from the EUPSA Registry. Eur J Pediatr Surg. 2025 Oct 15. doi: 10.1055/a-2708-2852[↩][↩]
- Velasco-Sandoval F, Santamaría-Díaz CE, Jiménez-López KI, Pais-Díaz F. VACTERL association in a newborn as a product of a twin pregnancy by egg donation. Cir Cir. 2025;93(3):315-319. English. doi: 10.24875/CIRU.22000128[↩][↩]
- Ávila MC, Rojas CM. Asociación VACTERL. Presentación de un caso en sesión anatomopatológica y consideraciones generales. Act Pediatr Mex. 2017;38:330-6. https://www.cirugiaycirujanos.com/frame_eng.php?id=1127&l=en[↩][↩]
- Ilyas E, Affan M, Iftikhar R, Abbasher Hussien Mohamed Ahmed K. A familial case of VACTERL association with co-occurring sacrococcygeal teratoma: a case report. Oxf Med Case Reports. 2025 May 28;2025(5):omaf046. doi: 10.1093/omcr/omaf046[↩][↩]
- Chaisrisawadisuk S, Tunkijjaroen T, Sathienkijkanchai A, Vatanavicharn N, Moore MH. VACTERL Association and Unilateral Lambdoid Craniosynostosis. Cleft Palate Craniofac J. 2025 Oct 3:10556656251383401. doi: 10.1177/10556656251383401[↩][↩]
- VACTERL Association. https://rarediseases.org/rare-diseases/vacterl-association/[↩][↩]
- VACTERL Association (VATER syndrome). https://www.chop.edu/conditions-diseases/vacterl-association-vater-syndrome[↩][↩]
- VACTERL Association (VATER Syndrome). https://www.nationwidechildrens.org/conditions/vacterl-association[↩][↩]
- VATER Syndrome/VACTERL Association. https://www.cincinnatichildrens.org/health/v/vacterl[↩][↩]
- VATER Syndrome (VACTERL Association). https://my.clevelandclinic.org/health/diseases/24083-vater-syndrome[↩][↩][↩]
- Chaisrisawadisuk S, Tunkijjaroen T, Sathienkijkanchai A, Vatanavicharn N, Moore MH. VACTERL Association and Unilateral Lambdoid Craniosynostosis. The Cleft Palate Craniofacial Journal. 2025;0(0). doi:10.1177/10556656251383401[↩][↩]
- Papadakis JE, Weber D, Albanese JS, Birch AK, Warf B. Incidence of idiopathic syrinx in pediatric patients diagnosed with VACTERL association. J Neurosurg Pediatr. 2025 Jan 10;35(4):385-390. doi: 10.3171/2024.10.PEDS24242[↩]
- Braungart S, Daff C. Expect the unexpected: neuroblastoma in a patient with the VACTERL association. BMJ Case Rep. 2025 Apr 8;18(4):e265123. doi: 10.1136/bcr-2025-265123[↩]
- Quan L, Smith DW. The VATER association. Vertebral defects, Anal atresia, T-E fistula with esophageal atresia, Radial and Renal dysplasia: a spectrum of associated defects. J Pediatr. 1973 Jan;82(1):104-7. doi: 10.1016/s0022-3476(73)80024-1[↩]
- Quan L, Smith DW. In: The clinical delineation of birth defects. Volume XII. G.I. tract including liver and pancreas. Bergsma D, editor. Baltimore: The Williams and Wilkins company; 1972. The VATER association: Vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, radial dysplasia; pp. 75–78.[↩]
- Temtamy SA, Miller JD. Extending the scope of the VATER association: definition of the VATER syndrome. J Pediatr. 1974 Sep;85(3):345-9. doi: 10.1016/s0022-3476(74)80113-7[↩]
- Solomon BD. VACTERL/VATER Association. Orphanet J Rare Dis. 2011 Aug 16;6:56. doi: 10.1186/1750-1172-6-56[↩][↩][↩][↩][↩][↩]
- Opitz JM. The developmental field concept. Am J Med Genet. 1985 May;21(1):1-11. doi: 10.1002/ajmg.1320210102[↩]
- Martínez-Frías ML, Frías JL, Opitz JM. Errors of morphogenesis and developmental field theory. Am J Med Genet. 1998;76:291–296. doi: 10.1002/(SICI)1096-8628(19980401)76:4<291::AID-AJMG3>3.0.CO;2-T[↩]
- Diaz-Frias J, Kondamudi NP. Alagille Syndrome. [Updated 2023 Aug 12]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507827[↩]
- Van Maldergem L, Piard J, Larizza L, et al. Baller-Gerold Syndrome. 2007 Aug 13 [Updated 2018 Apr 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1204[↩][↩][↩][↩]
- Debeljak M, Zver A, Jazbec J. A patient with Baller-Gerold syndrome and midline NK/T lymphoma. Am J Med Genet A. 2009 Feb 15;149A(4):755-9. doi: 10.1002/ajmg.a.32736[↩][↩]
- Usman N, Sur M. CHARGE Syndrome. [Updated 2023 Mar 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559199[↩]
- CHARGE syndrome. https://medlineplus.gov/genetics/condition/charge-syndrome/[↩]
- CHARGE Syndrome. https://rarediseases.org/rare-diseases/charge-syndrome/[↩]
- Currarino syndrome with two synchronous presacral teratomas. Journal of Pediatric Surgery Case Reports Volume 36, September 2018, Pages 16-18. https://www.sciencedirect.com/science/article/pii/S2213576618301507[↩][↩]
- Costanzo S, Spaccini L, Pio L, Mattioli G, Virgone C, Dall’Igna P, Iacobelli B, Inserra A, Brisighelli G, Fagnani AM, Leva E, Giannotti G, Cheli M, Frumento P, Riccipetitoni G. Currarino syndrome: does the presence of a genetic anomaly correlate with a more severe phenotype? A multicentre study. J Pediatr Surg. 2017 Oct;52(10):1591-1596. doi: 10.1016/j.jpedsurg.2017.06.012[↩][↩]
- Isik N, Elmaci I, Gokben B, Balak N, Tosyali N. Currarino triad: surgical management and follow-up results of four [correction of three] cases. Pediatr Neurosurg. 2010 Aug;46(2):110-9. doi: 10.1159/000319007. Epub 2010 Jul 20. Erratum in: Pediatr Neurosurg. 2010 Aug 46(2):150.[↩][↩]
- Baalaan KP, Gurunathan N. Currarino triad. Pan Afr Med J. 2022 Feb 17;41:143. doi: 10.11604/pamj.2022.41.143.33419[↩][↩]
- Fanconi anemia. https://medlineplus.gov/genetics/condition/fanconi-anemia[↩][↩]
- Bhandari J, Thada PK, Killeen RB, et al. Fanconi Anemia. [Updated 2024 Jun 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK559133[↩]
- Krauser AF, Ponnarasu S, Schury MP. Holt-Oram Syndrome. [Updated 2023 Aug 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513339[↩]
- Holt-Oram syndrome. https://medlineplus.gov/genetics/condition/holt-oram-syndrome/[↩]
- Holt-Oram syndrome: A case reportSíndrome Holt-Oram: a propósito de um caso clinic. Revista Portuguesa de Cardiologia (English Edition) Volume 33, Issue 11, November 2014, Pages 737.e1-737.e5 https://www.sciencedirect.com/science/article/pii/S2174204914002682[↩]
- Alasmari BG, Rayees S, Althubaiti S, Elzubair L, Chendeb S. Gray platelet syndrome in a neonate with VACTERL association: a novel homozygous pathogenic variant c.5257C>T in the NBEAL2 gene. Cureus. (2023) 15:e48359. 10.7759/cureus.48359[↩]
- Pangli BK, Braddock SR, Knutsen AP. Omenn syndrome in a 10-month-old male with athymia and VACTERL association. J Allergy Clin Immunol Glob. (2023) 2:100153. 10.1016/j.jacig.2023.100153[↩]
- Buttle SG, McMillan HJ, Davila J, Bokhaut J, Kovesi T, Katz SL, et al. Respiratory failure in a patient with VACTERL association and concomitant spinal muscular atrophy. Pediatr Pulmonol. (2023) 58:3314–9. 10.1002/ppul.26657[↩]
- Solomon BD. The etiology of VACTERL association: Current knowledge and hypotheses. Am J Med Genet C Semin Med Genet. 2018 Dec;178(4):440-446. doi: 10.1002/ajmg.c.31664[↩][↩]
- Reutter H, Hilger AC, Hildebrandt F, Ludwig M. Underlying genetic factors of the VATER/VACTERL association with special emphasis on the “Renal” phenotype. Pediatr Nephrol. 2016 Nov;31(11):2025-33. doi: 10.1007/s00467-016-3335-3[↩][↩]
- Salinas-Torres VM, Pérez-García N, Pérez-García G. Clinical, cytogenetic, environmental and inheritance findings in Mexican neonates with VACTERL association. Indian J Pediatr. 2015 Jan;82(1):84-8. doi: 10.1007/s12098-014-1493-5[↩][↩]
- Prevalence charts and tables. https://eu-rd-platform.jrc.ec.europa.eu/eurocat/eurocat-data/prevalence_en[↩]
- Kim P.C., Mo R., Hui Cc C. Murine models of VACTERL syndrome: Role of sonic hedgehog signaling pathway. J. Pediatr. Surg. 2001;36:381–384. doi: 10.1053/jpsu.2001.20722[↩]
- Friedland-Little J.M., Hoffmann A.D., Ocbina P.J.R., Peterson M.A., Bosman J.D., Chen Y., Cheng S.Y., Anderson K.V., Moskowitz I.P. A novel murine allele of Intraflagellar Transport Protein 172 causes a syndrome including VACTERL-like features with hydrocephalus. Hum. Mol. Genet. 2011;20:3725–3737. doi: 10.1093/hmg/ddr241[↩]
- Taschner M., Lorentzen E. The Intraflagellar Transport Machinery. Cold Spring Harb. Perspect. Biol. 2016;8:a028092. doi: 10.1101/cshperspect.a028092[↩]
- Wheway G., Nazlamova L., Hancock J.T. Signaling through the Primary Cilium. Front. Cell Dev. Biol. 2018;6:8. doi: 10.3389/fcell.2018.00008[↩]
- Tonni G, Koçak Ç, Grisolia G, Rizzo G, Araujo Júnior E, Werner H, et al. Clinical presentations and diagnostic imaging of VACTERL association. Fetal Pediatr Pathol. (2023) 42:651–74. 10.1080/15513815.2023.2206905[↩][↩][↩]
- Harjai MM, Holla RG, Kale R. Full spectrum of VACTERL in new born. Med J Armed Forces India. (2008) 64:84–5. 10.1016/s0377-1237(08)80163-3[↩]
- Harjai MM, Holla RG, Kale R. Full Spectrum of VACTERL in New Born. Med J Armed Forces India. 2008 Jan;64(1):84-5. doi: 10.1016/S0377-1237(08)80163-3[↩]
- Al-Farqani A, Panduranga P, Al-Maskari S, Thomas E. VACTERL Association with double-chambered left ventricle: a rare occurrence. Ann Pediatr Cardiol. (2013) 6:200–1. 10.4103/0974-2069.115283[↩]
- Yuan Z. Clinical analysis and literature review of one case with neonatal VACTERL association Hebei (Master’s thesis). Hebei Medical University, China(Hebei) (2019).[↩]
- Oral A, Caner I, Yigiter M, Kantarci M, Olgun H, Ceviz N, et al. Clinical characteristics of neonates with VACTERL association. Pediatr Int. (2012) 54:361–4. 10.1111/j.1442-200X.2012.03566.x[↩]
- Kimsey KM, Barnett GS, Keup C, Nguyen J, Wilsey MJ, Smithers CJ, et al. Esophageal heterotopic pancreas in an asymptomatic 2-year-old with VACTERL association. JPGN Rep. (2023) 4:e350. 10.1097/pg9.0000000000000350[↩]
- Yoon Y, Kim K, Yeom SK, Lee J, Lee Y. A case report of intrahepatic bile duct confluence anomalies in VACTERL syndrome. Medicine (Baltimore). (2018) 97:e12411. 10.1097/md.0000000000012411[↩]
- Solomon BD, Bear KA, Kimonis V, de Klein A, Scott DA, Shaw-Smith C, et al. Clinical geneticists’ views of VACTERL/VATER association. Am J Med Genet A. (2012) 158a:3087–100. 10.1002/ajmg.a.35638[↩]
- Massey H, Tennant S, Dean J; DDD study. PACS2, PACS1, and VACTERL: A Clinical Overlap. Mol Syndromol. 2025 Feb;16(1):29-32. doi: 10.1159/000539473[↩]
- Galarreta CI, Hoyt E, Forero L, Curry CJ, Bird LM. Ear anomalies and hearing loss in patients with VACTERL association and the effect of maternal diabetes. Am J Med Genet A. (2023) 191:2693–702. 10.1002/ajmg.a.63382[↩]
- Wang Z, Sun Z, Diao Y, Wang Z, Yang X, Jiang B, et al. Identification of two novel SALL1 mutations in Chinese families with townes-brocks syndrome and literature review. Orphanet J Rare Dis. (2023) 18:250. 10.1186/s13023-023-02874-4[↩]
- Graziano C, Olivucci G. SALL1-Related Townes-Brocks Syndrome. 2007 Jan 24 [Updated 2025 Aug 14]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1445[↩]
- Costa F, Valentim M, Ferreira C, Santos M. Navigating the anesthetic challenges of vertebral defects, anorectal anomalies, cardiac anomalies, tracheoesophageal Fistula (TEF)/esophageal atresia, renal anomalies, and limb abnormalities (VACTERL) association: a delicate balancing act. Cureus. (2024) 16:e68797. 10.7759/cureus.68797[↩]
- Raam MS, Pineda-Alvarez DE, Hadley DW, Solomon BD. Long-term outcomes of adults with features of VACTERL association. Eur J Med Genet. 2011 Jan-Feb;54(1):34-41. doi: 10.1016/j.ejmg.2010.09.007[↩][↩]
- Al-Naimi A, Hamad SG, Zarroug A. Outcome of newborns with tracheoesophageal Fistula: an experience from a rapidly developing country: room for improvement. Pulm Med. (2022) 2022:6558309. 10.1155/2022/6558309[↩]
- Kassa AM, Dahl M, Strinnholm M, Engstrand Lilja H. Attention difficulties and physical dysfunction common in children with complex congenital malformations: a study of preschool children with VACTERL association. Acta Paediatr. 2020 Apr;109(4):783-789. doi: 10.1111/apa.14566[↩]
- Bergmann S, Ritz LA, Widenmann-Grolig A, Jechalke S, von Schweinitz D, Hubertus J, et al. Swallowing-related quality of life in children with oesophageal atresia: a national cohort study. Eur J Pediatr. (2023) 182:275–83. 10.1007/s00431-022-04677-4[↩][↩]
- van Hoorn CE, de Graaff JC, Vlot J, Wijnen RM, Stolker RJ, Schnater JM. Primary repair of esophageal atresia is followed by multiple diagnostic and surgical procedures. J Pediatr Surg. (2021) 56:2192–9. 10.1016/j.jpedsurg.2021.06.004[↩]





{kind=link}