Contents
What is scleroderma
Scleroderma also known as systemic sclerosis is an autoimmune disease where your immune system attacks healthy tissue affecting connective tissue (the tissue that connects joints, muscles, blood vessels and internal organs) in your body 1. In scleroderma, the connective tissue gets hard or thick. Scleroderma can cause swelling or pain in your muscles and joints. Scleroderma involves overproduction of a protein called collagen in connective tissue. This results in hardening of the skin (scleroderma means hard skin). The hallmark of scleroderma is hardening of the skin. Some forms of scleroderma can also damage your blood vessels and internal organs.
Scleroderma is not contagious, infectious, cancerous or malignant. Scleroderma has no cure, but symptoms and damage can be reduced with proper treatment.
It’s estimated that about 300,000 Americans have scleroderma. About one third of those people have the systemic form of scleroderma. Scleroderma affects women more often than men (female patients outnumber male patients about 4-to-1) and most commonly occurs between the ages of 30 and 50. However, scleroderma can develop in every age group from infants to the elderly. Black people often have earlier onset and are more likely to have more skin involvement and lung disease.
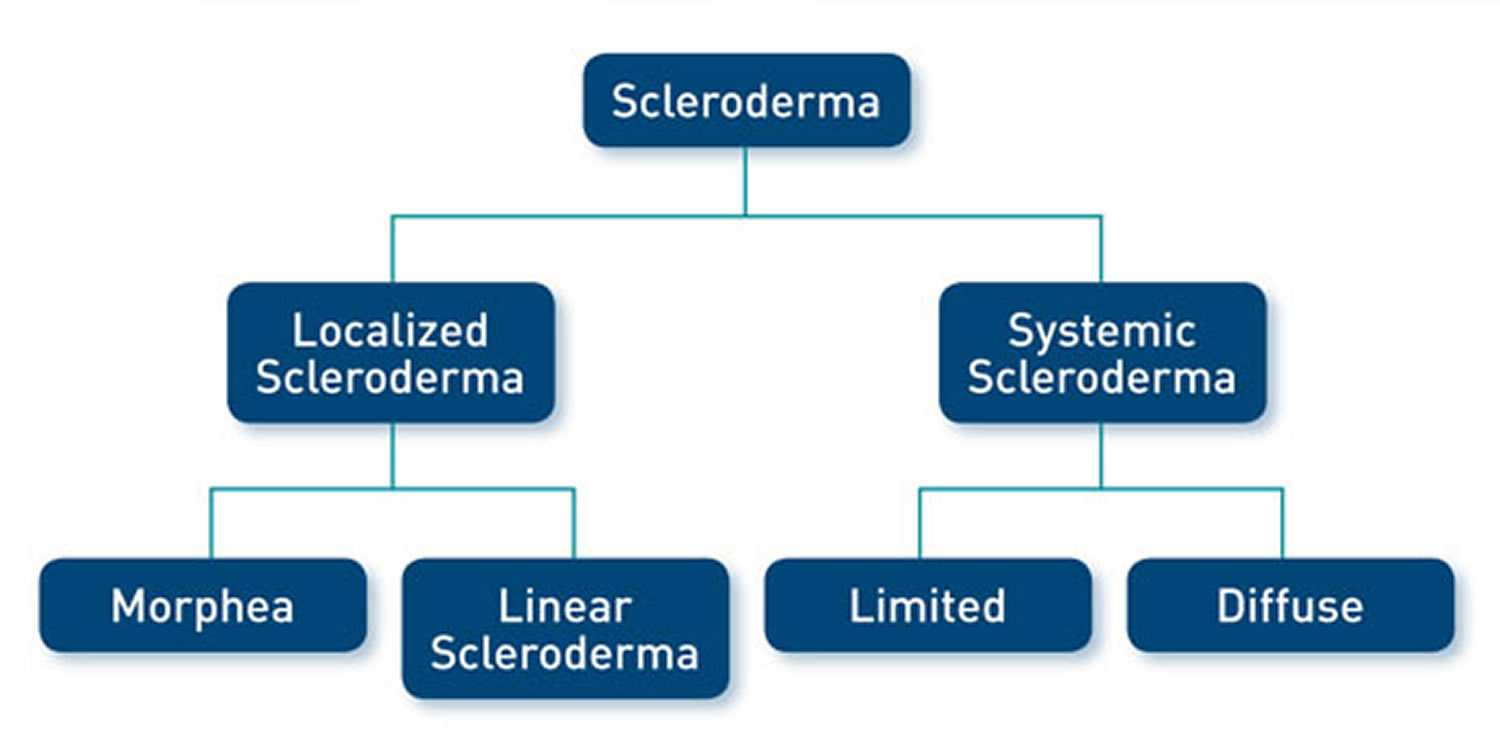
There are two types of scleroderma 1:
- Localized scleroderma also known as morphea, which only affects skin is more common in children. It does not harm major organs. It may get better or go away without help. But it can be severe in some people and can leave skin damage. Most localized scleroderma show up before age 40. They are also more common in people of European descent.
- Diffuse scleroderma also known as systemic sclerosis, which affects internal organs or blood vessels as well as skin. Systemic scleroderma are more common in people ages 30 to 50 and in African Americans.
The symptoms of scleroderma vary according to which part of your body is affected and it varies greatly for each person. Thickening and hardening of the skin is typical, especially on the fingers, arms and face. A mild scleroderma can become more serious if not properly treated. Prompt and proper diagnosis and treatment by qualified physicians may minimize the symptoms of scleroderma and lessen the chance for irreversible damage.
Another common symptom is Raynaud’s phenomenon, which is a blood circulation problem that causes your fingers or toes to change color and feel numb or painful in the cold. Primary Raynaud’s develops without any underlying condition whereas secondary Raynaud’s is linked to underlying disease, such as scleroderma.
Other symptoms of scleroderma include joint pain and stiffness, fatigue, indigestion or heartburn. Diffuse scleroderma can also cause symptoms related to the heart, lungs and kidneys.
There is currently no cure for scleroderma. However, treatment can improve symptoms. The treatment for scleroderma depends on what part of your body it is affecting. Your doctor may recommend stretching exercises for your joints, creams for your skin, dietary changes, or treatments to make the red patches on your skin less apparent.
Medication can improve blood circulation, and suppress the immune system, which may slow disease progress. If your organs are affected, you may be referred to a specialist, such as a kidney specialist if your kidneys are affected.
Lifestyle changes may make it easier to live with scleroderma. These include:
- wearing gloves and socks to keep your hands and feet warm, to prevent Raynaud’s phenomenon
- avoiding cigarette smoke, as this affects blood circulation
- regular physical activity to help keep skin and joints flexible
- keeping skin moisturized and clean to prevent dryness and infection
Figure 1. Scleroderma
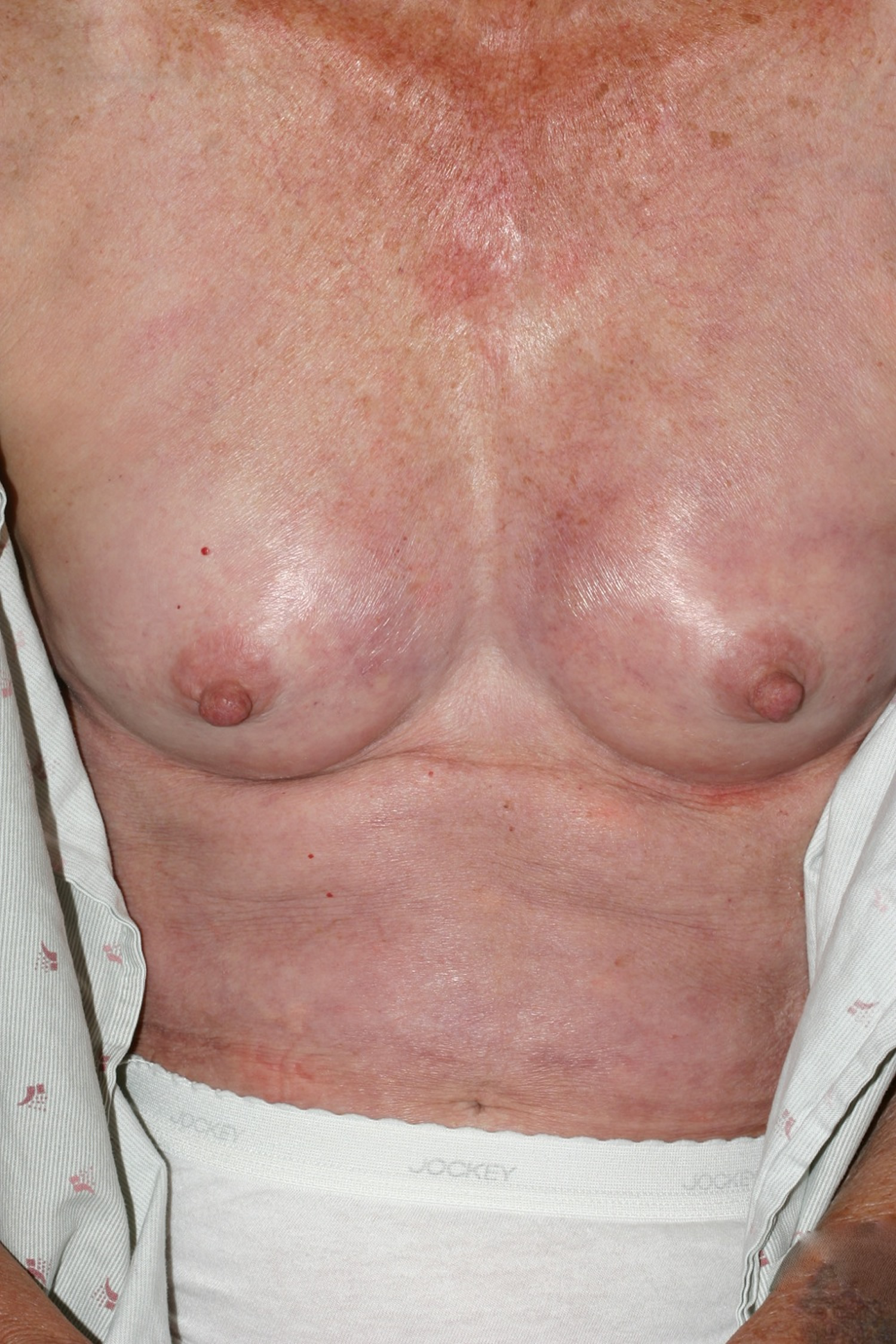
Figure 2. Limited scleroderma (salt and pepper depigmentation in systemic scleroderma)
Figure 3. CREST scleroderma
Localized Scleroderma
The changes, which occur in localized scleroderma, are usually found in only a few places on the skin or muscles, and rarely spread elsewhere. Generally, localized scleroderma is relatively mild. The internal organs are usually not affected, and persons with localized scleroderma rarely develop systemic scleroderma. Some laboratory abnormalities commonly seen in systemic scleroderma are frequently absent in the localized form.
Localized scleroderma is an uncommon condition, and affects approximately 20 people in every million. Although uncommon, localized scleroderma occurs approximately 20 times more often than systemic scleroderma. Both the incidence of morphea and linear scleroderma are more common in females, with the male to female ratio being approximately 3 to 4: 1. Plaque morphea is the most common form of localized scleroderma. However in childhood linear scleroderma is the most common.
Morphea
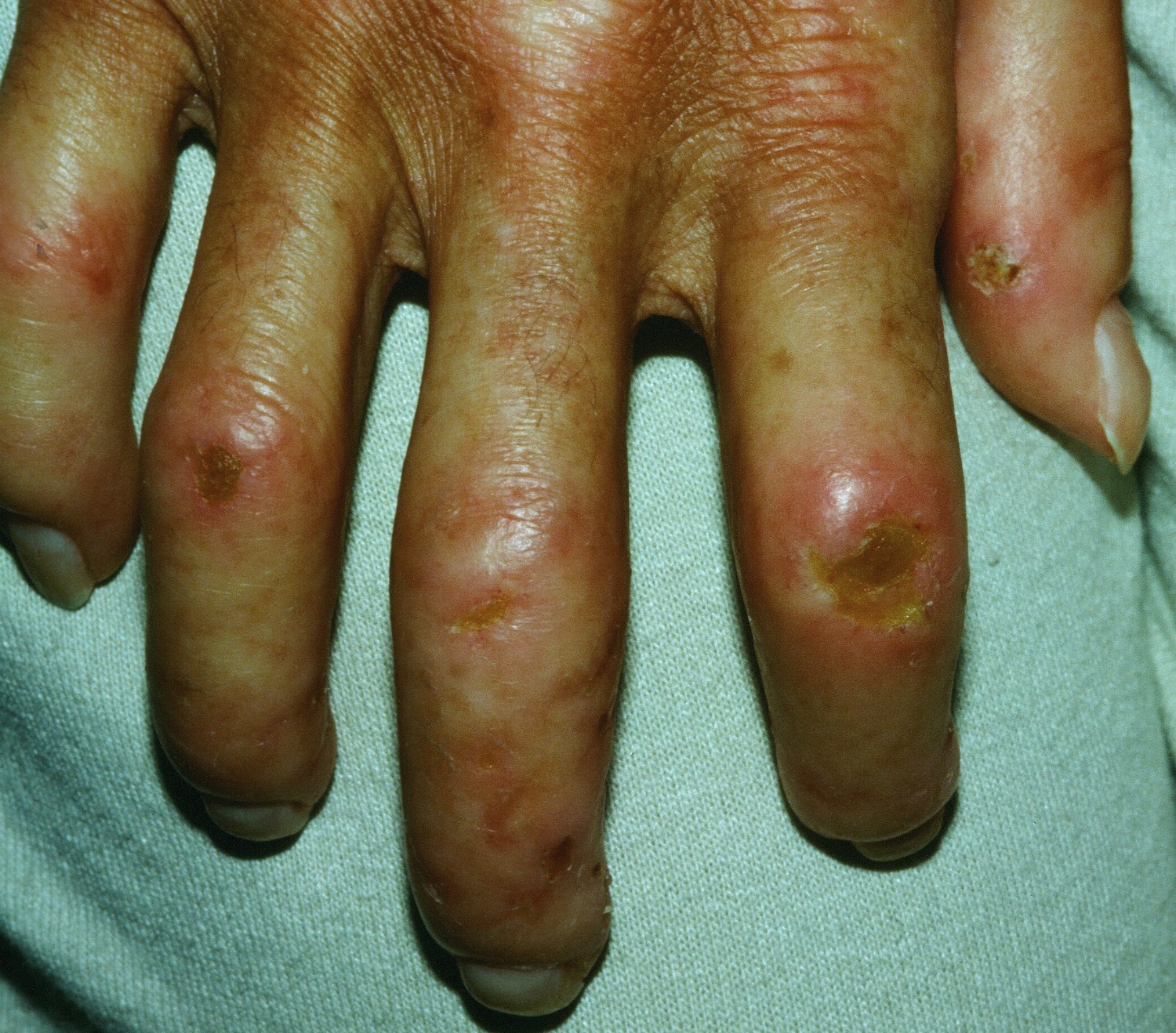
Morphea is a form of localized scleroderma characterized by waxy patches on the skin of varying sizes, shapes and color. Symptoms may start with reddish patches of skin that thicken into hardened, oval-shaped areas. The patches later become whitish in the middle with purple borders. Patches usually appear on the chest, stomach and back, but can also affect the face, arms and legs. People who have this type of scleroderma may have 1 or more patches that can be as small as half an inch and as large as 12 inches in diameter. The patches may enlarge or shrink, and often may disappear spontaneously. Morphea usually appears between the ages of 20 and 50, but is often seen in young children.
Generalized Morphea
In this form of localized scleroderma the lesions may gather in a single area or involve three or more areas. Common sites where these lesions are seen are on the trunk and the legs. Occasionally the acral (fingers, toes and ears) areas of the patient’s body are spared. This condition can cause deep involvement of tissues causing disfigurement.
Figure 4. Morphea
Linear scleroderma
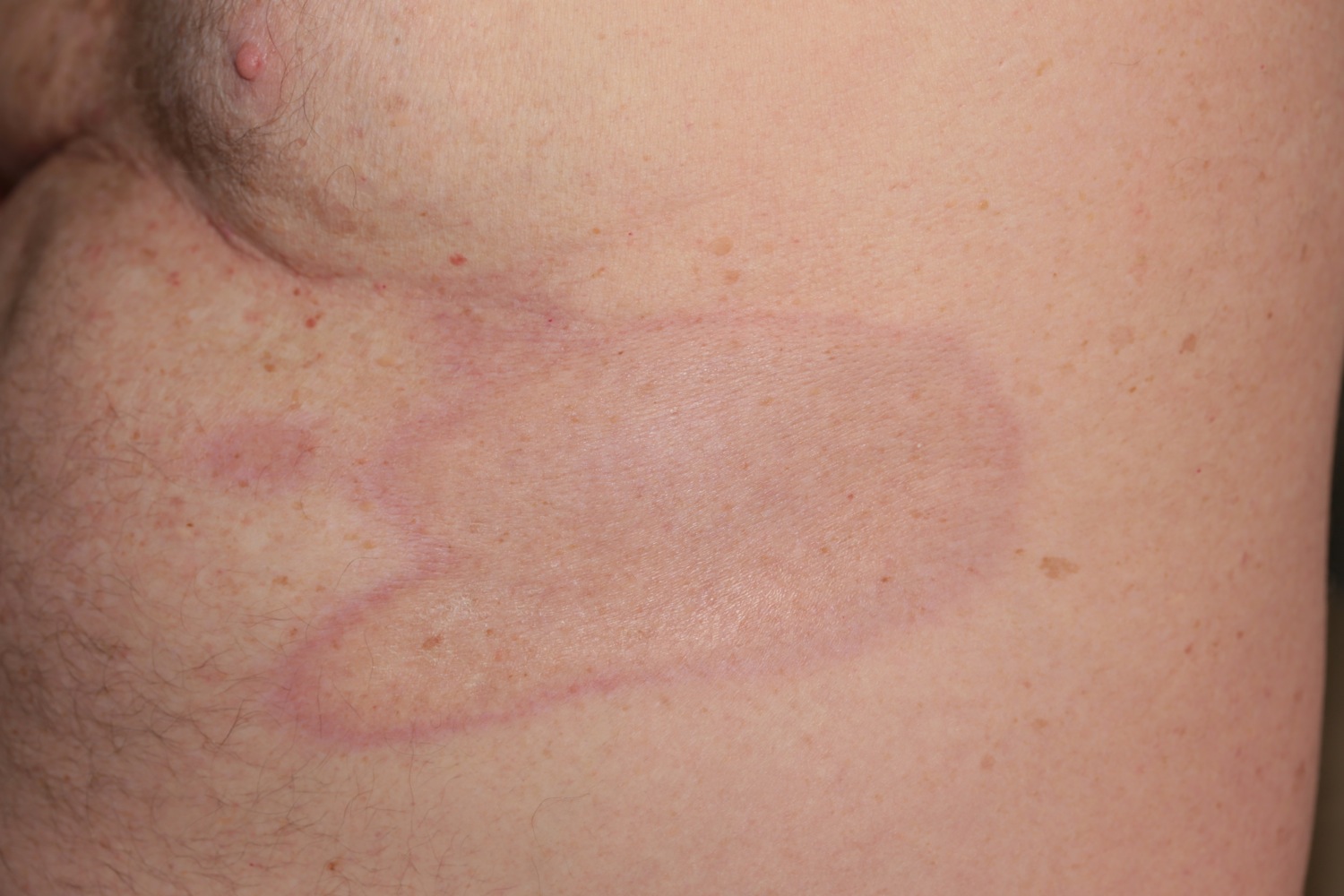
Linear scleroderma is a form of localized scleroderma which frequently starts as a streak or line of hardened, waxy skin on an arm or leg or on the forehead. Sometimes it forms a long crease on the head or neck, referred to as en coup de sabre because it resembles a saber or sword wound. Linear scleroderma tends to involve deeper layers of the skin as well as the surface layers, and sometimes affects the motion of the joints, which lie underneath. Linear scleroderma usually develops in childhood. In children, the growth of involved limbs may be affected. In 10 to 22% of cases of linear scleroderma there is shortening of the affected limbs due to impaired growth.
There are many complications of linear scleroderma, these can include the following:
- Tissue destruction that can involve: soft tissue, muscle, bone and sometimes the lining of joints.
- Defects in growth in different areas of the limb
- Deformities of the limb
- Involvement of the skin surrounding the mouth and spread of lesions into the oral cavity causing severe dental problems such as the premature loss of primary teeth, eruption of the permanent teeth and reduced growth of bone.
Linear scleroderma en coup de sabre
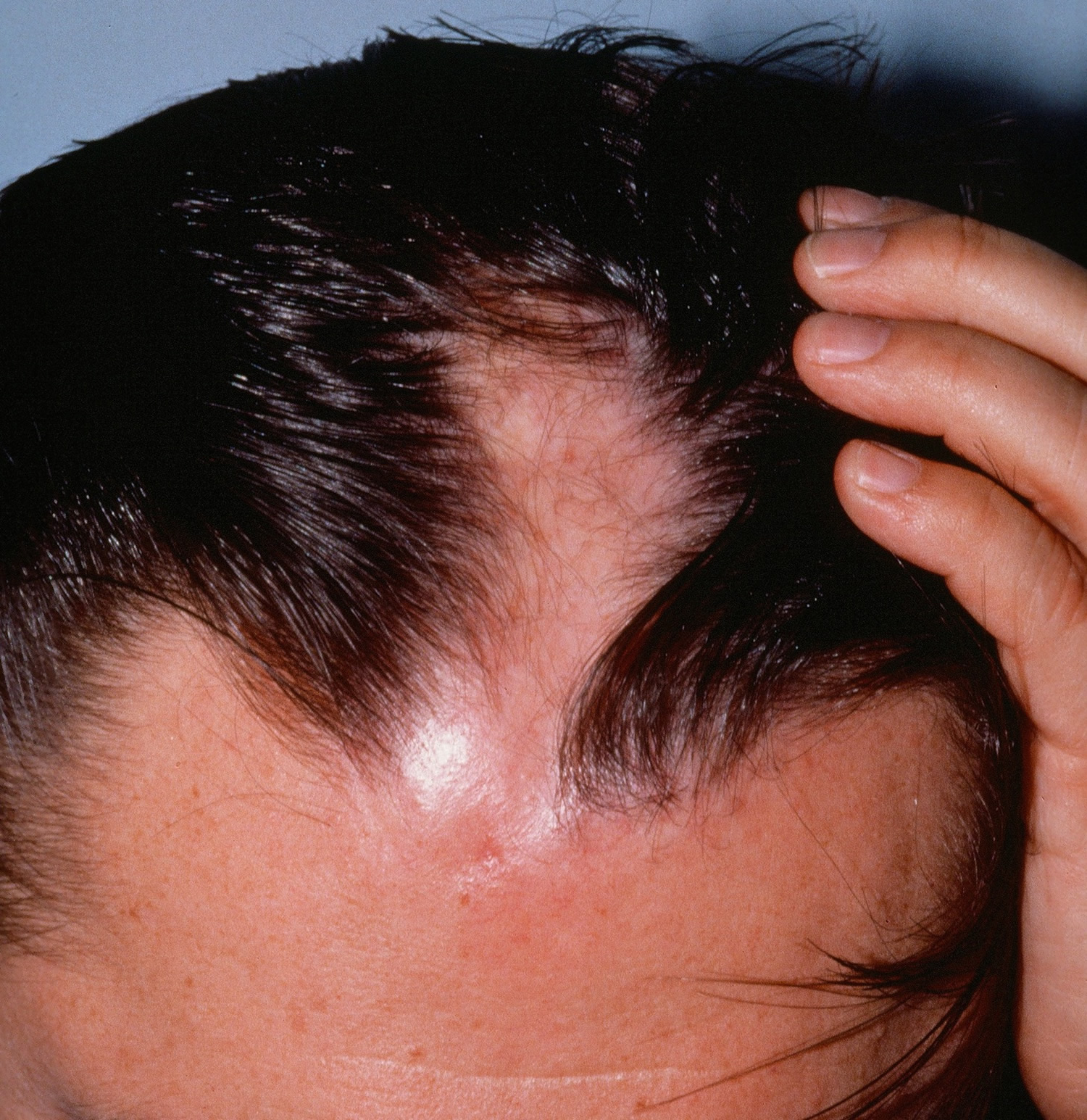
Linear scleroderma en coup de sabre is the term generally applied when children have linear scleroderma on the head or face. This form of linear scleroderma describes lesions that resemble a scar from a sabre blow to the head. Originally, this term applied to children with a deep furrow along the scalp with tight, hard skin that often extended to the forehead.
Children with linear scleroderma en coup de sabre are a very diverse group. Some children appear to have the classically described disease with lesions only on the scalp and forehead, while other children may have lesions only on the chin or lip. There is another group of children who are termed as having Parry Romberg syndrome. Children with this condition have similar skin lesions, but may have involvement of the whole side of the face and even involvement of the tongue. Obvious cases of en coup de sabre and Parry Romberg syndrome differ significantly, but many children present with crossover manifestations, which can make it difficult to determine with certainty, which form of scleroderma the child has.
The long-term outcome for children with linear scleroderma en coup de sabre is mixed. If the lesions are confined to the scalp and forehead, they often evolve similarly to linear scleroderma and the effect is primarily cosmetic. The same is true for isolated areas of involvement on the face. Parry Romberg syndrome, in which one side of the face is involved, can present additional serious difficulties because the bones may not grow properly.
Figure 5. Linear scleroderma en coup de sabre
Localized scleroderma treatment in children
Appropriate treatment for children with localized scleroderma depends on the area of skin involved. Small areas on the body may not need any therapy while lesions on the face or over the joints often are treated aggressively. For children with mild morphea, topical treatment with calcipotriene cream or ointment during the active phase is often sufficient. Linear scleroderma often responds well to methotrexate, and physicians may use it for children with widespread or otherwise disturbing morphea.
Linear scleroderma that does not cross a joint line and is not of sufficient extent to suggest it will cause deformity, probably does not require treatment. However, when large areas of an extremity are involved, or the disease is crossing a joint line, there is a significant risk of permanent damage. In these cases, most specialists will recommend therapy with methotrexate, which is often associated with a slow but steady softening of the involved skin. Like all medicines, methotrexate is not without possible side effects and careful consideration should be given to both the expected risks and benefits. The use of methotrexate for children with linear scleroderma en coup de sabre remains controversial. Discuss the benefits and risks of the medication with your child’s doctor. Proper therapy for children with Parry Romberg syndrome is unknown.
Although there are no scientific studies, many families have found the application of commercially available cocoa butter preparations useful. Many doctors suggest you avoid food additives or dietary supplements, as they can be potentially harmful.
Systemic scleroderma (systemic sclerosis)
The changes occurring in systemic scleroderma may affect the connective tissue in many parts of the body. Systemic scleroderma can involve the skin, esophagus, gastrointestinal tract (stomach and bowels), lungs, kidneys, heart and other internal organs. It can also affect blood vessels, muscles and joints. The tissues of involved organs become hard and fibrous, causing them to function less efficiently. The term systemic sclerosis indicates that “sclerosis” (hardening) may occur in the internal systems of the body. There are two major recognized patterns that the illness can take – diffuse or limited disease.
Overlap syndrome
Up to 20% of patients with systemic scleroderma have an overlap syndrome with another connective tissue disease and develop arthritis, lupus or myositis.
Systemic scleroderma and pregnancy
Women with systemic scleroderma may find it harder to get pregnant and could have a slightly higher risk of miscarriage and giving birth prematurely. However, if the pregnancy is planned in consultation with a doctor during a period when the condition is stable, there’s no reason why a woman with systemic scleroderma cannot have a successful pregnancy.
Systemic scleroderma is rare, with prevalence varying from 30–500 cases per million.
- It is up to 5 times more common in females compared to males.
- All races and ethnicities may be affected, but rates appear to be slightly higher in some Native Americans and black-skinned races, and lower in those of Asian background.
- Peak age of onset of systemic scleroderma varies between approximately 35 and 55 years. Juvenile onset occurs but is rare compared to adult onset disease.
Key features of systemic scleroderma
- Skin thickening of the fingers and toes (sclerodactyly)
- Specific autoantibodies in the blood (anti-Scl70 or anti-centromere antibody and others)
- Abnormal nail fold capillaries
- Internal organ fibrosis and/or vascular damage (involving the lungs, heart, gastrointestinal tract and/or kidneys)
Systemic scleroderma causes
Systemic scleroderma is an autoimmune condition characterized by inflammation, fibrosis and vasculopathy.
The precise underlying mechanisms are complex and remain largely unknown. Genetic susceptibility plus a triggering event result in a cascade of innate and adaptive immunoinflammatory responses.
Genetic susceptibility
- First degree relatives of affected individuals may be at 10–16 fold increased risk of developing systemic sclerosis, compared with those with no family history of the disease.
- Studies have identified genetic loci associated with systemic sclerosis.
- Clinical subtypes map to particular genetic subsets.
- Differences in gene expression occur in fibroblasts, immune (T and B), endothelial, smooth muscle and epithelial cells.
Triggering event
Systemic scleroderma has been associated with injury, exposure to silica, vinyl chloride monomer, chlorinated solvents, trichloroethylene, welding fumes, aromatic solvents, ketones, bleomycin and possibly other drugs (vitamin K, cocaine, penicillamine, appetite suppressants and some chemotherapeutic agents).
Immune pathways
A number of pathways are likely involved in the pathogenesis of systemic sclerosis, including cytokines that injure blood vessels, growth factors that stimulate collagen production, integrin signalling, morphogen pathways, co-stimulatory pathways and more.
Diffuse scleroderma
Two-thirds of patients with systemic sclerosis have diffuse systemic scleroderma: skin involvement is widespread and includes proximal limbs. In diffuse scleroderma, skin thickening occurs more rapidly and involves more skin areas than in limited disease. It can affect the skin all over the body, causing it to swell, become shiny, tight and itchy. Eventually, skin becomes soft again, and may go back to normal. In addition, people with diffuse scleroderma have a higher risk of developing “sclerosis” or fibrous hardening of the internal organs, causing damage to internal organs such as the intestines, lungs, kidneys and heart.
Limited scleroderma
About one-third of patients have a slower and more benign illness called limited scleroderma. Limited systemic scleroderma is limited to the digits, distal limbs (not spreading more proximal than the elbows or knees) and face. Limited systemic scleroderma progresses more slowly than diffuse systemic scleroderma and with less internal organ involvement except there is a risk of pulmonary artery hypertension, especially later in the disease course.
Although internal problems do occur, they are less frequent and tend to be less severe than in diffuse scleroderma, and are usually delayed in onset for several years. However, persons with limited scleroderma, and occasionally those with diffuse scleroderma, can develop pulmonary hypertension, a condition in which the lung’s blood vessels become narrow, leading to impaired blood flow through the lungs resulting in shortness of breath.
Limited scleroderma is sometimes called CREST syndrome. CREST stands for the initial letters of five common features:
- Calcinosis
- Raynaud Phenomenon
- Esophageal dysfunction
- Sclerodactyly
- Telangiectasia
To further complicate the terminology, some people with diffuse disease will go on to develop calcinosis and telangiectasias so that they also have the features of CREST.
Although most patients can be classified as having diffuse or limited disease, different people may have different symptoms and different combination of symptoms of the illness.
Figure 6. Raynaud’s phenomenon
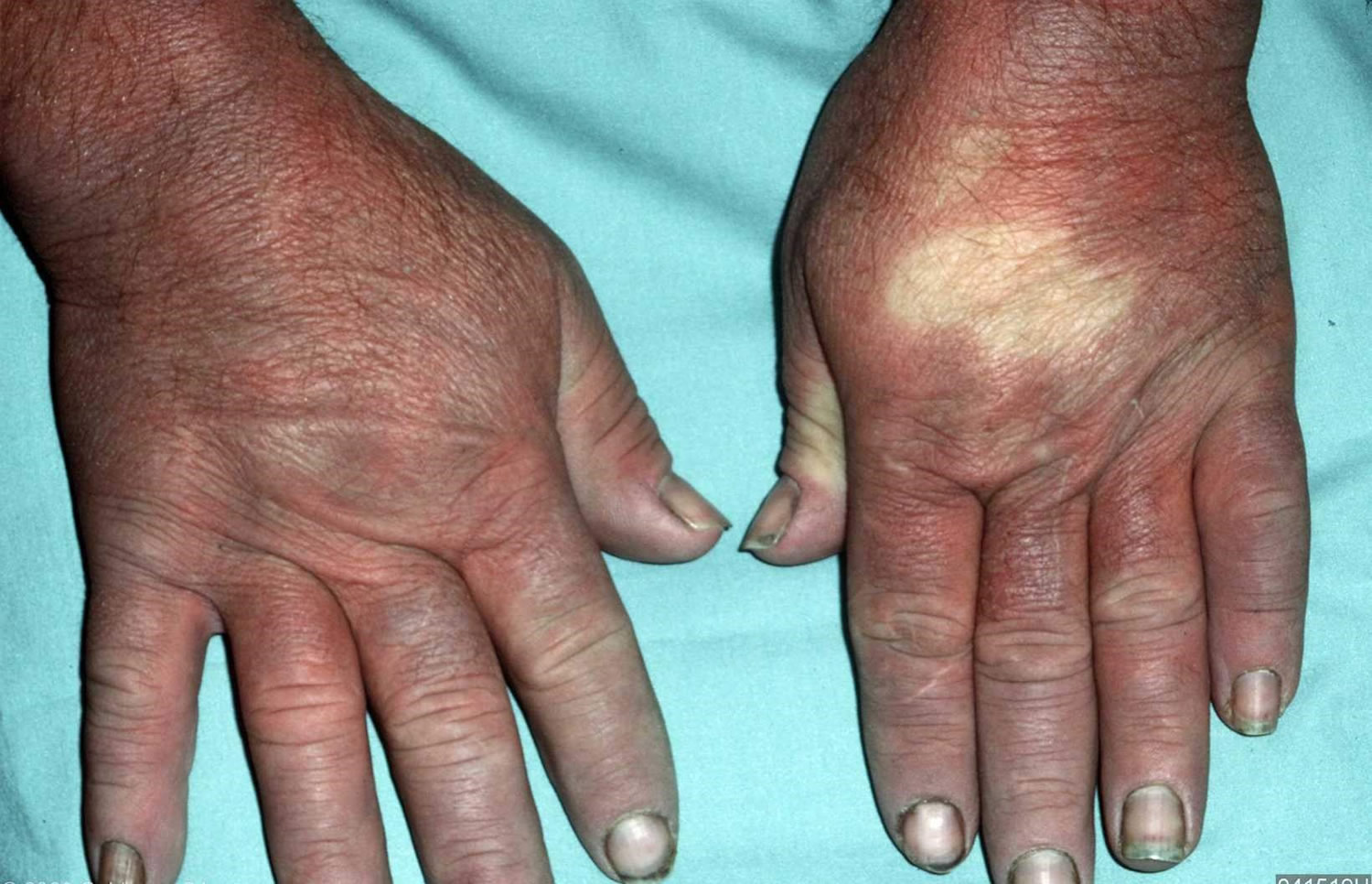
Figure 7. Sclerodactyly
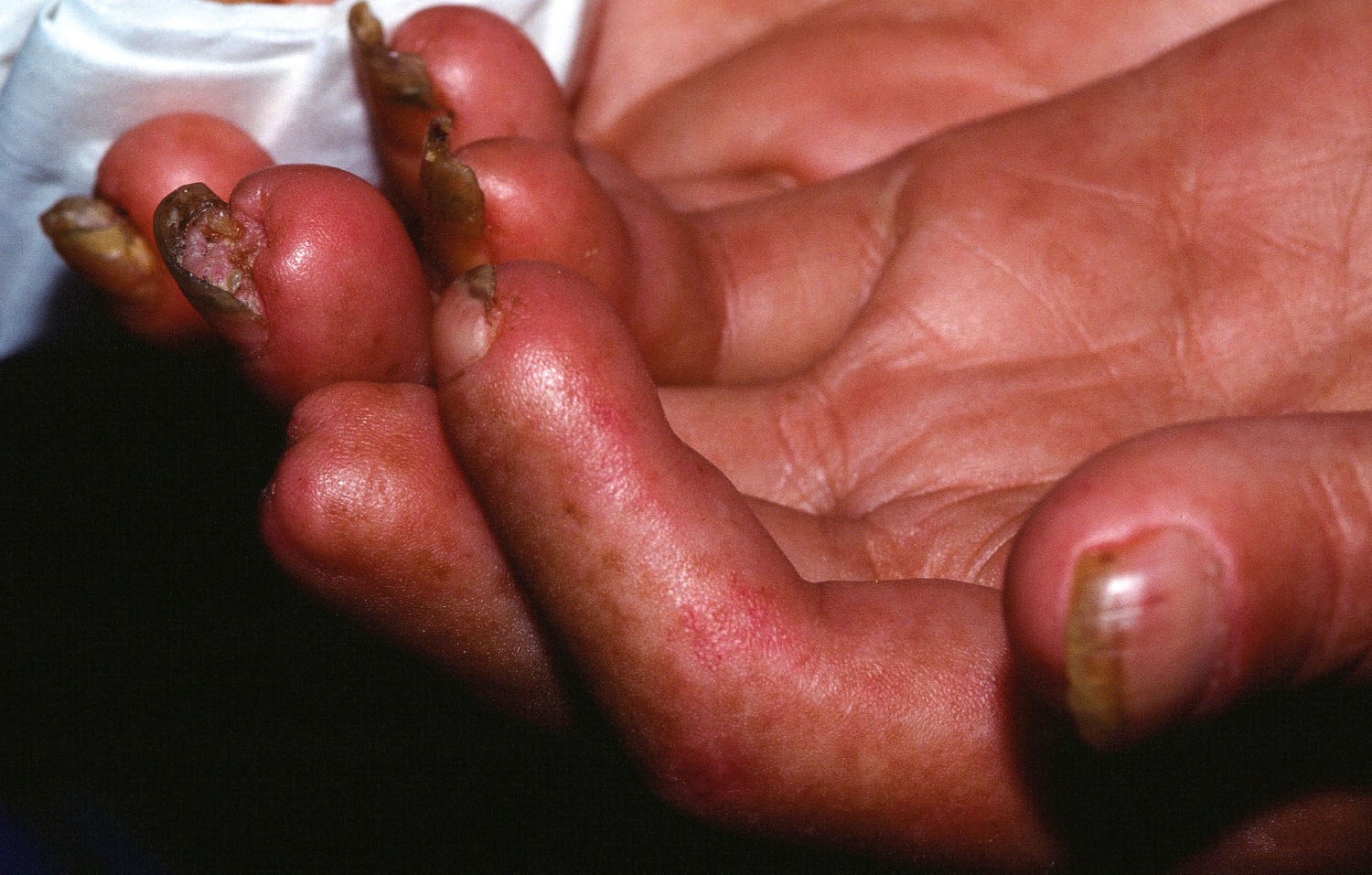
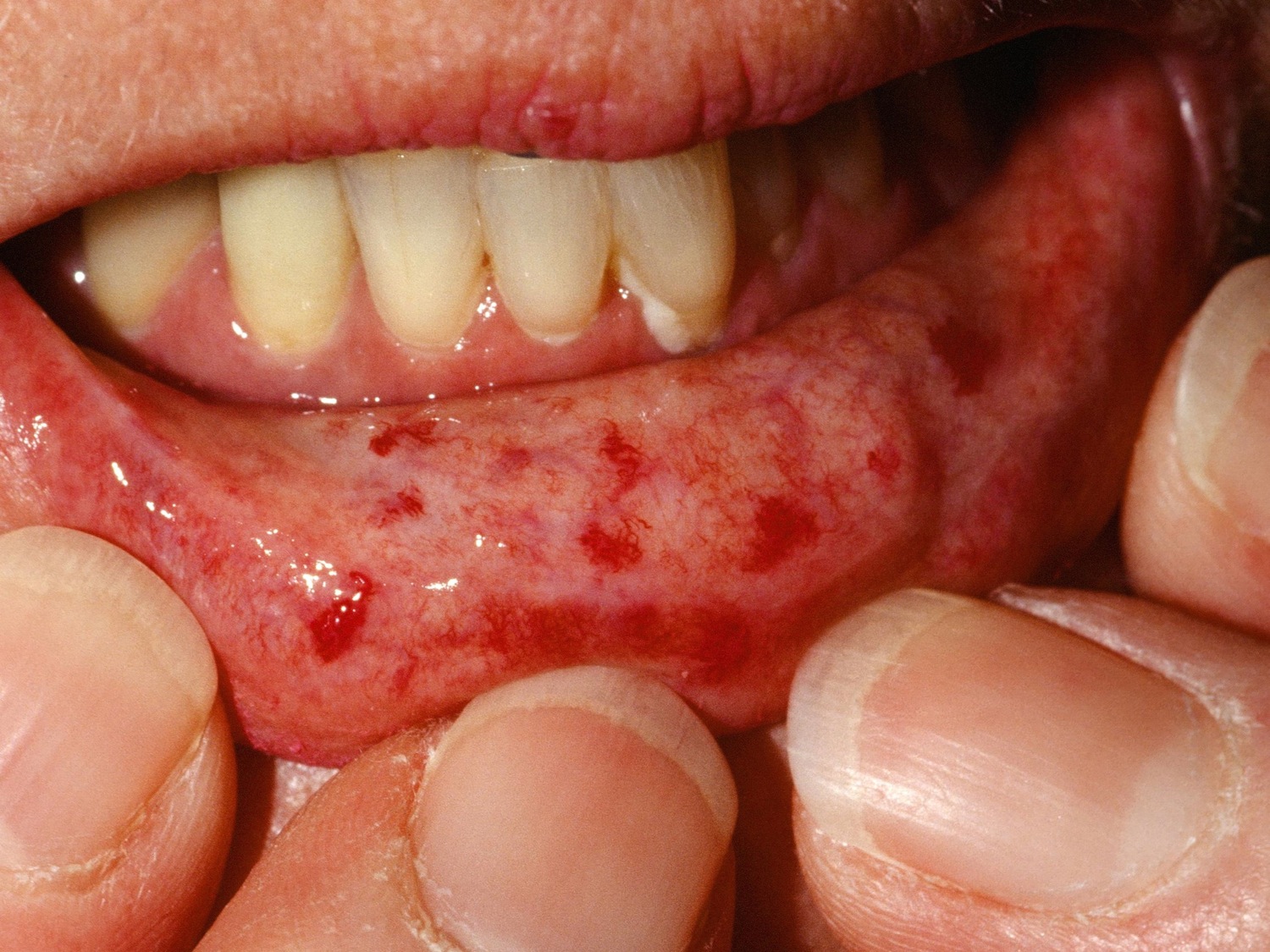
Figure 8. Telangiectasia
Scleroderma life expectancy
People with systemic scleroderma experience have a reduced life expectancy. The degree of skin disease as well as involvement of other organs is associated with a worse prognosis. Survival is determined by the disease subset and internal organ complications. Interstitial lung disease and pulmonary artery hypertension account for almost two-thirds of deaths related to systemic scleroderma. Pulmonary hypertension, pulmonary fibrosis (interstitial lung disease), and scleroderma renal crisis are the most frequent causes of death 2, 3, 4, 5, 6, 7, 8, 9.
Survival has improved in recent decades and correlates best with the clinical disease subtype (diffuse cutaneous vs limited cutaneous) and with the extent of organ involvement.
In localized scleroderma remission is common, in addition there is a very low chance that internal organ disease will occur. Existing data illustrates that there is a 50% chance of the lesions softening or remitting withing 3.8 years. In the plaque subtype of localized scleroderma the average duration of disease before spontaneous remission is 2.7 years. In the deep forms of the disease the average time before remission is 5.5 years.
Survival in patients with diffuse skin scleroderma has improved significantly; currently, the 5-year survival is estimated to be about 80% (from 69% in the 1990–1993 cohort to 84% in the 2000–2003 cohort). Five-year survival in patients with limited skin systemic sclerosis is approximately 90% and unchanged for the same periods (93% and 91%, respectively) 10.
Factors associated with a more severe scleroderma prognosis are as follows 10:
- Younger age
- African descent
- Rapid progression of skin symptoms
- Greater extent of skin involvement
- Anemia
- Elevated erythrocyte sedimentation rate (ESR)
- Lung, kidney, and heart involvement.
Cigarette smoking has been found to reduce overall survival 11. In a multicenter French cohort study, age older than 60 years at diagnosis, diffuse skin scleroderma, scleroderma renal crisis, dyspnea, 6-minute walking distance, forced vital capacity less than 70%, diffusing capacity of the lungs for carbon monoxide less than 70%, pulmonary hypertension, telangiectasia, valvular disease, malignancy, anemia, and C-reactive protein greater than 8 mg/L were linked with poorer survival 12.
Scleroderma prognosis
There is no cure for systemic scleroderma. Scleroderma is a chronic (long-lasting) disease and the prognosis depends on the type of systemic sclerosis. Although symptoms may come and go over time, the various forms of this disease usually last a lifetime. The skin swelling that happens first can last for a few weeks or months. This is followed by a gradual thickening of the skin and other skin changes. In the diffuse form of the disease, skin symptoms tend to peak within three years, then stabilize or even improve. If skin changes occur more rapidly, there is often a greater risk that internal organs are being damaged as well. In limited scleroderma, skin symptoms tend to worsen very slowly over a period of many years. Proactive and routine annual screening allows early intervention with disease modifying drugs. These have led to improvement in prognosis and long term outcomes in recent years.
In the limited form of systemic sclerosis, a patient’s condition can be stable for years 13. However, in diffuse systemic sclerosis, the disease can rapidly lead to death, if it is not treated promptly 13. Pulmonary hypertension may be an important cause of mortality in these patients. Survival complicated by pulmonary hypertension has been poor despite available treatment options. Pulmonary hypertension was documented 25% of elderly patients, yet only in 12% of the youngest ones in a large study that stressed the differences in early- versus late-onset systemic scleroderma 14.
For patients with scleroderma renal crisis, 1-year mortality is 20-30% and 5-year mortality is 30-50%; for normotensive scleroderma renal crisis, which accounts for about 10% of all cases, 1-year mortality is 60% 15. Half of patients with scleroderma require hemodialysis, and kidney function recovers spontaneously in only one quarter of those. Predictors of worse outcome in patients with scleroderma renal crisis include the following 16:
- Older age
- Male gender
- Serum creatinine > 3 mg/dL at presentation
- Incomplete blood pressure control within the first 3 days of the crisis
- Normal blood pressure
- Use of an angiotensin-converting enzyme (ACE) inhibitor prior to scleroderma renal crisis is also an independent predictor of poor outcome, possibly because by keeping the blood pressure under control, the medication blunts recognition of the unfolding renal crisis.
Mortality associated with scleroderma renal crisis has declined significantly in recent decades. In contrast, pulmonary involvement (interstitial lung disease and/or pulmonary arterial hypertension) has become the most common cause of death in patients with systemic sclerosis 10.
Scleroderma complications
Scleroderma complications range from mild to severe and can affect your:
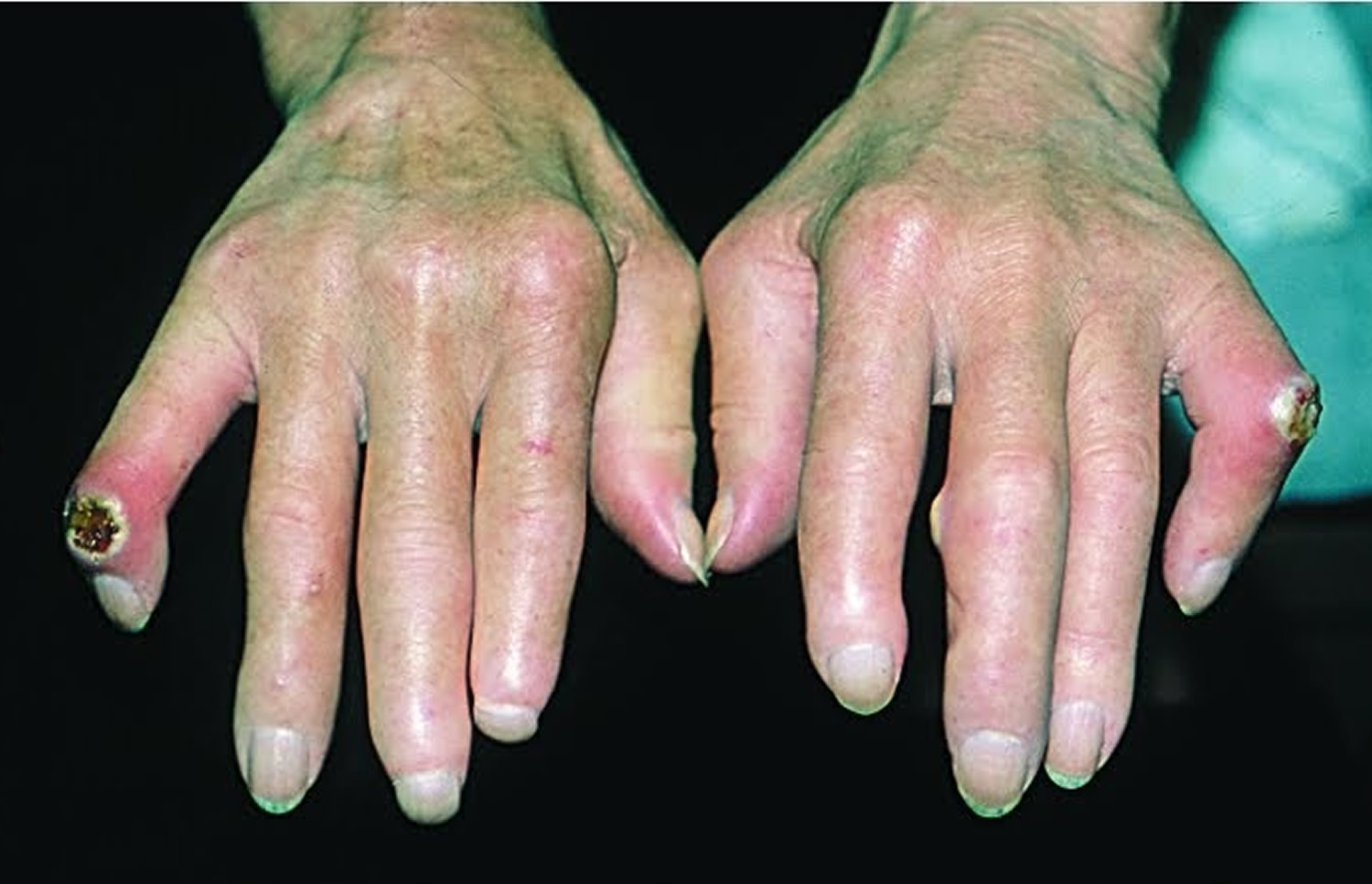
- Fingertips. The variety of Raynaud’s disease that occurs with scleroderma can be so severe that the restricted blood flow permanently damages the tissue at the fingertips, causing pits or skin sores (ulcers). In some cases, gangrene and amputation may follow.
- Joints. The skin over joints can become so tight that it restricts flexibility and movement, particularly in the hands.
- Lungs. Scarring of lung tissue (pulmonary fibrosis) can result in reduced lung function, reduced ability to breathe and reduced tolerance for exercise. You may also develop high blood pressure in the arteries to your lungs (pulmonary hypertension).
- Kidneys. When scleroderma affects your kidneys, you can develop elevated blood pressure and an increased level of protein in your urine. More-serious effects of kidney complications may include renal crisis, which involves a sudden increase in blood pressure and rapid kidney failure. Prompt treatment of this condition is important to preserve kidney function.
- Heart. Scarring of heart tissue increases your risk of abnormal heartbeats (arrhythmias) and congestive heart failure, and can cause inflammation of the membranous sac surrounding your heart (pericarditis). Scleroderma can also raise the pressure on the right side of your heart and cause it to wear out.
- Teeth. Severe tightening of facial skin can cause your mouth to become smaller and narrower, which may make it hard to brush your teeth or to even have them professionally cleaned. People who have scleroderma often don’t produce normal amounts of saliva, so the risk of dental decay increases even more.
- Digestive system. Digestive problems associated with scleroderma can lead to acid reflux and difficulty swallowing — some describe feeling as if food gets stuck midway down the esophagus — as well as bouts of constipation alternating with episodes of diarrhea. Some people who have scleroderma also may have problems absorbing nutrients due to overgrowth of bacteria in the intestine.
- Sexual function. Men who have scleroderma often experience erectile dysfunction. Scleroderma may also affect the sexual function of women, by decreasing sexual lubrication and constricting the vaginal opening. A cross-sectional study that included 90 women with systemic sclerosis and 90 healthy controls aged 18 to 70 years documented a high prevalence of impaired pelvic floor function and sexual function in women with systemic sclerosis 17. The prevalence of female sexual dysfunction was 73% in systemic sclerosis patients versus 31% in the controls 17. Impaired sexual function correlated with higher disease activity, the presence of dyspnea and interstitial lung disease, increased systemic inflammation, reduced physical activity, functional disability, more severe depression, more pronounced fatigue, and impaired quality of life 17.
Frerix and colleagues have proposed that osteonecrosis of the lunate bone (Kienböck disease), while very rare in the general population, may be a frequent manifestation of systemic sclerosis, and may be linked to vasculopathy from the disorder 18. Lunate osteonecrosis manifests as pain and stiffness in the affected wrist and decreased grip strength in the hand.
Scleroderma causes
The exact cause or causes of scleroderma are still unknown and it is believed that both genetic and environmental factors are thought to contribute to its development 1, 19. It is known that scleroderma results from an overproduction and accumulation of collagen in your body tissues. Collagen is a fibrous type of protein that makes up your body’s connective tissues, including your skin. Doctors aren’t certain what prompts this abnormal collagen production, but your body’s immune system appears to play a role. In some genetically susceptible people, symptoms may be triggered by exposure to certain types of pesticides, epoxy resins or solvents. Although scleroderma is not directly inherited, some scientists feel there is a slight predisposition to it in families with a history of rheumatic diseases.
In individuals with a genetic predisposition, environmental factors (eg, triggers or accelerators) may contribute to the development of systemic sclerosis 20, 21, 22, 23, 24. These environmental factors (eg, triggers or accelerators) include the following 25:
- Silica exposure
- Solvent exposure (vinyl chloride, trichloroethylene, epoxy resins, benzene, carbon tetrachloride)
- Radiation exposure or radiotherapy
Case reports describe the development of scleroderma in patients treated with the immune checkpoint inhibitor pembrolizumab for metastatic melanoma 26.
Cytomegalovirus (CMV), human herpesvirus 5, and parvovirus B19 have been proposed as viral accelerating factors. However, evidence of their involvement is inconclusive 27.
Several scleroderma-like disorders, which can be distinguished from systemic sclerosis by clinical, histopathological, and laboratory features, are associated with environmental exposures such as contaminated rapeseed cooking oil causing toxic oil syndrome and L-tryptophan linked to eosinophilia-myalgia syndrome 28. In addition, certain drugs, such as bleomycin and cocaine, have been associated with developing systemic sclerosis-like illnesses 1.
Is scleroderma genetic?
Systemic sclerosis is not inherited, although a genetic predisposition plays an important role in its development. Most patients do not have any relatives with scleroderma and their children do not get scleroderma 19. Research indicates that there is a susceptibility gene, which raises the likelihood of getting scleroderma, but by itself does not cause the disease.
Scleroderma is often associated with clusters of multiple other autoimmune diseases within families 29, 30. Genetic studies have confirmed the involvement of the major histocompatibility complex genetic region in systemic sclerosis, akin to other autoimmune disorders like systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) 1. The association of specific human leukocyte antigens (HLA), including HLA DRB1*1104, DQA1*0501, and DQB1*0301, with systemic sclerosis has been long known. Further, non-HLA loci such as PTPN22, NLRP1, STAT4, and IRF5 have also been implicated in the cause of systemic sclerosis 31.
Risk factors for scleroderma
Scleroderma occurs much more often in women than it does in men. Choctaw Native Americans and African-Americans are more likely than Americans of European descent to develop the type of scleroderma that affects internal organs.
Several environmental triggers are associated with the subsequent development of scleroderma, including infectious agents such as Cytomegalovirus (CMV), Epstein-Barr virus, and parvovirus B19 25. Exposure to silica dust is also associated with scleroderma, along with occasional exposure to other agents such as organic solvents, toluene, xylene, trichloroethylene, and polyvinyl chloride 1. Cigarette smoking is not a proven risk factor for scleroderma 1.
Several other combined factors appear to influence the risk of having scleroderma:
- Genetics. People who have certain gene changes appear to be more likely to develop scleroderma. This may explain why scleroderma appears to run in families in a small number of people and why some types of scleroderma are more common for people in certain racial and ethnic groups.
- Environmental triggers. Research suggests that in some people, scleroderma symptoms may be triggered by exposure to certain viruses, medicines or drugs. Repeated exposure, such as at work, to certain harmful substances or chemicals also may increase the risk of scleroderma. An environmental trigger is not identified for most people.
- Immune system conditions. Scleroderma is believed to be an autoimmune disease. This means that it occurs in part because the body’s immune system begins to attack the connective tissues. People who have scleroderma also may have symptoms of another autoimmune disease such as rheumatoid arthritis, systemic lupus erythematosus (SLE) or Sjogren syndrome.
Scleroderma symptoms
Scleroderma causes your tissues to get hard and thick. Depending on what type of scleroderma you have, you may find that your skin gets hard and tight, or you may have problems with your blood vessels and major organs, such as your heart, lungs, and kidneys.
Scleroderma’s signs and symptoms vary, depending on which parts of your body are involved:
- Skin. Nearly everyone who has scleroderma experiences a hardening and tightening of patches of skin. These patches may be shaped like ovals or straight lines, or cover wide areas of the trunk and limbs. The number, location and size of the patches vary by type of scleroderma. The first parts of the body to be affected are usually the fingers, hands, feet and face. In some people, the skin thickening also can involve the forearms, upper arms, chest, abdomen, lower legs and thighs. Early symptoms may include swelling and itchiness. The color of affected skin can become lighter or darker, and skin may look shiny because of the tightness. Skin can appear shiny because it’s so tight, and movement of the affected area may be restricted. Some people also have small red spots, called telangiectasia, on their hands and face. Calcium deposits can form under the skin, particularly at the fingertips, causing bumps that can be seen on X-rays.
- Fingers or toes. One of the earliest signs of scleroderma is an exaggerated response to cold temperatures or emotional distress, which can cause numbness, pain or color changes in the fingers or toes. Called Raynaud’s disease or Raynaud’s phenomenon, this condition also occurs in people who don’t have scleroderma.
- Raynaud’s disease also called Raynaud’s phenomenon is a rare disorder of the blood vessels, usually in your fingers and toes. Raynaud’s phenomenon causes the blood vessels to narrow when you are cold or feeling stressed. When this happens, blood can’t get to the surface of the skin and the affected areas turn turn white, blue, gray or red and your fingers and toes may feel painful or numb. When the blood flow returns, the skin turns red and throbs or tingles. In severe cases, loss of blood flow can cause sores or tissue death.
- Digestive system. Scleroderma can affect any part of your digestive system, from the esophagus to the rectum. In addition to acid reflux, which can damage the section of esophagus nearest the stomach, some people with scleroderma may also have problems absorbing nutrients if their intestinal muscles aren’t moving food properly through the intestines (e.g. malabsorption, repeated episodes of pseudo-obstruction, or severe problems requiring hyperalimentation – artificial supply of nutrients typically intravenously). Depending on which parts of your digestive system are affected, your symptoms may include:
- Heartburn
- Difficulty swallowing
- Bloating
- Diarrhea
- Constipation
- Fecal incontinence.
- Heart, lungs or kidneys. Scleroderma can affect the function of your heart (e.g. cardiomyopathy, symptomatic pericarditis, or arrhythmia), lungs (e.g. pulmonary fibrosis) or kidneys (e.g. “renal crisis”, kidney failure) to varying degrees. These problems, if left untreated, can become life-threatening.
- When scleroderma affects the heart or lungs, it can cause shortness of breath, decreased exercise tolerance and dizziness. Scleroderma can cause scarring in the lung tissues that may result in increasing shortness of breath over time. There are medicines that may help slow the progression of this lung damage.
- Scleroderma also can cause the blood pressure to increase in the circulation that goes between the heart and the lungs. This is called pulmonary hypertension. In addition to causing shortness of breath, pulmonary hypertension also can cause excess fluid to build up in the legs, feet and sometimes around the heart.
- When scleroderma affects the heart, heartbeats can become irregular. Heart failure also may happen in some people.
- Other symptoms. Other symptoms can include dry eyes and mouth, sudden episodes of severe facial pain (trigeminal neuralgia), and impotence.
More than 95% of people with scleroderma have both Raynaud’s phenomenon and skin thickening also called sclerodactyly when the fingers are involved. In addition, those with limited scleroderma tend to have telangiectasias, a collection of dilated blood vessels under the skin (85% of patients); digestive problems involving the esophagus (80%); and calcinosis (50%), often called CREST syndrome (calcinosis, Raynaud’s, esophageal disease, sclerodactyly, and telangiectasia). High pressure in the blood vessels around the lung a serious condition called pulmonary hypertension develops in about 15% of people with limited scleroderma.
Besides having Raynaud’s phenomenon and skin thickening, people with the diffuse form of scleroderma can have digestive symptoms involving the esophagus (80%), joint symptoms (70%), muscle weakness (50%), lung symptoms (40%), and heart failure (30%).
Clinical features of localized scleroderma
Morphea
Plaque Morphea
In this condition there is reddening of the skin with swelling. As the disease goes on the skin turns white and sunken. It most commonly occurs on the trunk but can also occur on the extremities. The size of the lesions rages from 0.5cm to 30cm. Later in the disease the lesions may become overly pigmented or stay under-pigmented. In lesions that are progressing a surrounding area of redness is seen called the ‘lilac ring’. There is rarely ever internal organ involvement.
Guttate Morphea
This condition usually produces multiple and small ‘confetti’ like lesions, 2 to 10mm in diameter. The lesions are under-pigmented or over-pigmented spots with a thin overlying skin. This condition frequently involves the shoulders and the chest.
Morphea Profundus
In this condition the skin becomes thickened and tighter. The skin loses it color and becomes sunken in.
Nodular (Keloidal) Scleroderma
This condition most commonly involves the chest and appears as distinct papules (small areas of elevated skin) or plaques (large areas of elevated skin). They are difficult to distinguish from keloids (an abnormally aggressive healing response). These lesions can be many centimeters in diameter, there may be single or multiple lesions.
Bullous Morphea
The formation of bullae (fluid filled lesions) can occur in most types of localized scleroderma. Common sites where the lesions can appear are on the extremities, trunk, face, or neck. These lesions may be superficial or extend deep into the skin. It is postulated that the bullae form in response to trauma or lymphatic obstruction.
Vesicular Morphea
In localized scleroderma vesicles (smaller fluid filled lesions) can develop, these are most often seen on the arms.
Atrophoderma of Pasini and Pierini
Lesions in this condition are described as being depressed, over-pigmented areas of skin, usually on the trunk, with a ‘cliffdrop’ border. This condition can occur alone or with other forms of localized scleroderma.
Clinical features of systemic scleroderma
The clinical features of systemic scleroderma are related to underlying vascular, inflammatory and fibrotic disease. Constitutional symptoms are common, such as fatigue, arthralgia and myalgia.
Cutaneous features of systemic scleroderma
Skin scleroderma
- Extent of skin fibrosis defines diffuse vs limited systemic scleroderma
- Sclerodactyly: thickening and tightness of the skin of the fingers (or toes). May be spindle-shaped
Hands
- Puffy fingers; early inflammatory phase of disease
- Raynaud phenomenon
- Abnormal nail fold capillaries
- Palmar erythema affecting thenar / hypothenar eminence
- Smaller fragile nails with ragged cuticles
- Digital pitted scars
- Digital ulcers
- Ulceration can lead to dry gangrene and eventual loss of the tips of the fingers (like frost bite).
Face
- Matt telangiectases on face, chest, palms
- Peri-oral furrowing (fat loss)
- Microstomia (limited oral aperture defined as interlabial distance < 4.5 cm)
- Beaked nose
Other
- Calcinosis affecting digits, extensor surfaces of limbs. Skin can breakdown and discharge chalky material (calcium)
- Salt and pepper dyspigmentation (hyperpigmentation and hypopigmentation)
- Pruritus
Rarer cutaneous features
- Morphoea
- Most frequently plaque, nodular or linear
- More common in limited cutaneous systemic scleroderma
- Panniculitis
Gastrointestinal symptoms
Upper Gastrointestinal Tract
- Gastroesophageal reflux / heart burn
- Dyspepsia
- Dysphagia
- Early satiety
- Micro-aspiration (accelerates lung disease)
Lower Gastrointestinal Tract
- Bloating, distention
- Nausea, vomiting
- Pain
- Alternating diarrhea / constipation
- Incontinence (anorectal sphincter dysfunction)
Cardiopulmonary symptoms
- Interstitial lung disease
- Pulmonary artery hypertension
- Cardiac scleroderma (cardiomyopathy, conductive)
- Shortness of breath
- Decreased exercise tolerance
- Chest pain
- Palpitations
Renal disease
Scleroderma renal crisis:
- Proteinuria
- High blood pressure
- Renal failure.
Other symptoms
- Fatigue
- Sicca symptoms (dry eyes, dry mouth) and Sjogren syndrome
- Musculoskeletal symptoms: friction rubs over the joints and tendons, particularly the knees; joint pain, muscle pain, weakness and limited movement resulting in contractures
- Ocular symptoms: tight eyelids, reduced tear secretion, retinopathy
Scleroderma diagnosis
Because scleroderma can take so many forms and affect so many different areas of your body, it can be difficult to diagnose. The diagnostic process may require consultation with rheumatologists (arthritis specialists), and/or dermatologists (skin specialists) and require blood studies and numerous other specialized tests depending upon which organs are affected.
Your doctor will ask about your symptoms and will examine your skin, especially on your fingers, hands, and face. If your doctor suspects you have scleroderma, he or she may want to order blood tests to check for elevated blood levels of certain antibodies produced by the immune system and urine tests. Occasionally, a skin biopsy may be recommended, during which a small sample of skin is removed and examined in a laboratory. If scleroderma affects internal organs such as the heart, lungs, or digestive organs, a chest x-ray and other tests may be necessary.
Your doctor may also suggest breathing tests (pulmonary function tests), a CT scan of your lungs and an echocardiogram of your heart.
Autoantibody Tests
Autoantibodies are an essential diagnostic tool, offering predictive insights into disease phenotype and prognosis of scleroderma. Antinuclear antibodies (ANA), detected through direct immunofluorescence, are positive in over 90% of systemic sclerosis cases 32. In instances where antinuclear antibodies (ANA) are negative, it is important to rule out other potential diagnoses before confirming systemic sclerosis. Specific autoantibodies, with a positivity rate of 60% to 70%, offer further diagnostic specificity. These antibodies, as mentioned below, which are mutually exclusive, can manifest several months to years before the clinical onset of systemic sclerosis.
- Anti-centromere antibody: Anti-centromere antibodies target 4 centromere antigens (CENP -B, -A, -C, D) and are predominantly observed in limited cutaneous systemic sclerosis, although they may occasionally occur in diffuse cutaneous systemic sclerosis. They can also manifest in conditions such as Sjögren syndrome and systemic lupus erythematosus. Anti-centromere antibodies are associated with an increased risk of pulmonary arterial hypertension (PAH). Furthermore, they are linked to limited skin involvement, a reduced likelihood of interstitial lung disease (ILD), and generally better survival rates compared to other autoantibodies.
- Anti-topoisomerase I (Scl-70) antibody: Anti-topoisomerase I (Scl-70) antibodies target the catalytic region of DNA helicase topoisomerase I. They are predominantly observed in diffuse cutaneous systemic sclerosis and infrequently in limited cutaneous systemic sclerosis. The presence of anti–Scl-70 antibodies is linked to an elevated risk of diffuse cutaneous involvement, interstitial lung disease, and heart involvement 33.
- Anti-RNA polymerase III antibody: Anti-RNA polymerase III antibodies target the eukaryotic RNA polymerase III. They are particularly associated with diffuse cutaneous systemic sclerosis and are linked to rapidly progressing and aggressive diffuse skin involvement, poor cutaneous outcomes, and scleroderma renal crisis. Additionally, these antibodies are also associated with a lower risk of interstitial lung disease and pulmonary arterial hypertension (PAH). Some studies have indicated an association between anti-RNA polymerase III antibodies and malignancies in systemic sclerosis patients.
- Anti–U3-RNP (fibrillarin) antibody: Anti-U3-RNP (fibrillarin) antibodies are prevalent in African Americans and are associated with an overall poor prognosis in systemic sclerosis. Their presence correlates with increased internal organ involvement, diffuse cutaneous manifestations, interstitial lung disease, pulmonary arterial hypertension (PAH), scleroderma renal crisis, myositis/myopathy, and heart complications 34.
- Other autoantibodies: Other autoantibodies include anti-Th/To antibodies that are associated with limited skin disease. Anti–PM/Scl antibodies associated with limited skin disease and overlap syndrome predispose to inflammatory myositis and interstitial lung disease. Anti–U1-RNP antibodies are more prevalent in African Americans and are associated with overlap syndrome and mixed connective tissue disease. This antibody profile is linked to limited cutaneous involvement and increased risk of inflammatory arthritis, myositis, lupus skin rashes, and lupus nephritis. Anti-Ku antibodies are also associated with overlap syndrome in systemic sclerosis, which is associated with more inflammatory arthritis and myositis.
Serological and genetic classification
Different autoantibody profiles are associated with particular clinical features and prognosis, particularly the pattern of antinuclear antibody (ANA) reactivity. Genetic associations in systemic scleroderma can also be mapped to particular ANA subtypes.
Centromere (kinetochore) ANA pattern
- Limited cutaneous systemic scleroderma
- Pulmonary artery hypertension
- Digital ulcers
- Calcinosis
- Associated genes: HLA-DQB1, TNF-863A, NOTCH4
Pm-Scl (nucleolar) ANA pattern
- Myositis overlap
U1RNP (speckled) ANA pattern
- Overlap with features of other connective tissue diseases
- Associated gene: HLA-DQB1
Th-To (nucleolar) ANA pattern
- Limited cutaneous systemic scleroderma
- Pulmonary artery hypertension
- Interstitial lung disease
- Poor prognosis
Topoisomerase-1 / Scl-70 (speckled) ANA pattern
- Diffuse cutaneous systemic scleroderma
- Interstitial lung disease
- Cardiac scleroderma
- Associated genes: HLA-DPA1, HLA-DRB1
RNA polymerase (P) III (fine speckled, nucleolar) ANA pattern
- Diffuse cutaneous systemic scleroderma
- Scleroderma renal crisis
- Malignancy
- Pulmonary artery hypertension
- Associated genes: HLA-DRB1, HLA-DRB3, HLA-DRB4, EDNRA
Fibrillarin / U3RNP (nucleolar)
- Afro-Caribbean asssociation
- Pulmonary artery hypertension
- Myositis
- Cardiac scleroderma
- Gastrointestinal involvement
- Associated gene: HLA-DQB1
Systemic scleroderma diagnostic criteria
The diagnosis of systemic scleroderma is confirmed when key features are present.
- Sclerodactyly
- Abnormal nail folds capillaries on capillaroscopy/dermatoscopy
- Specific autoantibodies (especially anti-Scl70 or anti-centromere antibody)
- Internal organ fibrosis and/or vascular damage
Investigations may include:
- Other blood tests: anaemia, raised sedimentation rate (ESR) and C-reactive protein (CRP), positive Rheumatoid factor, increased gamma globulins (hypergammaglobulinaemia) and abnormal coagulation tests may be present.
- Skin biopsy: excessive ground substance and odd-looking endothelial cells in the dermis; and later, deposits of collagen. The epidermis is usually atrophic (thinned).
- When muscle involvement is suspected, electromyography (EMG) or nerve conduction velocity testing is the initial diagnostic choice. If abnormal results are obtained, a muscle biopsy may be warranted.
- Pulmonary function tests. Pulmonary function tests, including spirometry, lung volumes, and diffusion capacity, can detect a restrictive pattern in interstitial lung disease very early in the disease process. A lung biopsy is of very limited value and should be avoided unless other diagnostic concerns are concurrent. When pulmonary arterial hypertension (PAH) is suspected, transthoracic echocardiography is usually initially performed, although right heart catheterization should follow to confirm the diagnosis and rule out other causes. Electrocardiography and Holter monitoring can be useful in detecting arrhythmias.
- High resolution CT scan. High-resolution CT (HRCT) scan is the preferred imaging modality for assessing interstitial lung disease, as it can detect subtle findings that may not be visible on a chest x-ray 35.
- Echocardiogram
- Right heart catheter
- ECG
- Cardiac MRI. In patients suspected of myocardial involvement, a cardiac MRI may be necessary.
- Barium swallow, manometry
- Endoscopy with gastrointestinal biopsy. When esophageal and upper gastrointestinal involvement is suspected, upper gastrointestinal endoscopy, esophageal manometry, barium swallow studies, and a 24-hour pH probe can be used 36. A characteristic finding indicative of esophageal dysmotility is a dilated esophagus with excessive air observed on a CT scan.
The joint American College of Rheumatology and European League against Rheumatism classification criteria (2013) are utilized to diagnose systemic scleroderma 37. A score of 9 or more confirms the diagnosis.
- Skin thickening of the fingers of both hands extending proximal to metacarpal phalangeal joint: score 9
- Skin thickening of the fingers (only count the highest score)
- Puffy fingers: score 2
- Sclerodactyly of the fingers (between distal and proximal interphalangeal joints): score 4
- Finger tip lesions (only count the highest score)
- Fingertip pitting scars: score 3
- Digital tip ulcers: score 2
- Telangiectasia: 2
- Telangiectasia(e) in a scleroderma like pattern are round and well demarcated and found on hands, lips, inside of the mouth, and/or large matt-like telangiectasia(e). Telangiectasiae are visible macular dilated superficial blood vessels; which collapse upon pressure and fill slowly when pressure is released; distinguishable from rapidly filling spider angiomas with central arteriole and from dilated superficial vessels.
- Abnormal nailfold capillaries (enlarged capillaries and/or capillary loss with or without peri-capillary hemorrhages at the nailfold and may be seen on the cuticle): 2
- Raynaud phenomenon: 3
- Lung disease (Maximum score of 2)
- Pulmonary artery hypertension (PAH): 2
- Interstitial lung disease (ILD): 2
- Positive systemic sclerosis-specific antibodies (Maximum score is 3)
- Any of anti-centromere or anti-topoisomerase I (also known as anti-Scl70 antibody): 3
- Anti–Scl–70, and anti–RNA polymerase III: 3
Scleroderma Differential Diagnosis
Scleroderma is primarily a clinical diagnosis. However, several other diseases that can mimic systemic sclerosis should be considered when establishing a differential diagnosis.
- Eosinophilic Fasciitis: Eosinophilic fasciitis is characterized by eosinophilic inflammation of the deep fascia, resulting in thickening and a woody induration of the upper and lower extremities, excluding the hands and feet 38. Raynaud phenomenon is not associated with eosinophilic fasciitis, and nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are generally negative in eosinophilic fasciitis cases. Patients may develop contractures resembling those seen in systemic sclerosis. Eosinophilic fasciitis may be associated with underlying malignancies. Histologically, a skin biopsy often reveals eosinophilic infiltrates in the deep fascia 38, 39.
- Scleromyxedema: Scleromyxedema is usually seen in patients with monoclonal gammopathy or multiple myeloma and is characterized by papular waxy lesions on the face, neck, extremities, and fingers 40. Patients may also exhibit associated symptoms such as seizures and dementia. Raynaud phenomenon is not a feature of scleromyxedema, and nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are typically negative in scleromyxedema cases. Histologically, a skin biopsy of scleromyxedema often reveals dermal fibrosis accompanied by perivascular inflammation and the deposition of mucin and fibrocytes, which are not typical findings in systemic sclerosis 40.
- Scleredema (scleroedema): Scleredema (scleroedema) can be associated with conditions such as diabetes mellitus, monoclonal gammopathy, fatigue, infections, and cancers 41. This condition is characterized by the doughy, indurated appearance of the skin on the neck, back, and face, with digits typically being spared. Raynaud phenomenon is not associated with scleredema (scleroedema), and the nailfold capillary examination appears usually normal. Antinuclear antibodies (ANA) and specific autoantibodies are generally negative in scleredema cases. Histologically, a skin biopsy of scleredema (scleroedema) often reveals dermal fibrosis without perivascular inflammation, along with mucin deposition 41.
- Nephrogenic Systemic Fibrosis: Nephrogenic systemic fibrosis is a rare phenomenon observed in patients with end-stage renal disease following exposure to gadolinium contrast. This condition is characterized by cobblestone-like nodular plaques on the extremities, trunk, hands, and feet while sparing the face. Raynaud phenomenon is not associated with nephrogenic systemic fibrosis, and the nailfold capillary examination typically appears normal. Antinuclear antibodies and specific autoantibodies are generally negative in nephrogenic systemic fibrosis cases. Histologically, a skin biopsy of nephrogenic systemic fibrosis often reveals dermal and epidermal fibrosis without perivascular inflammation, accompanied by the deposition of mucin and fibrocytes.
- Eosinophilia Myalgia Syndrome: Eosinophilia Myalgia Syndrome was an epidemic associated with the use of L-tryptophan, resulting in severe myalgias, visceral involvement, and elevated creatine kinase (CK) levels.
- Toxic Oil Syndrome: Toxic Oil Syndrome was an epidemic in Spain associated with the consumption of adulterated rapeseed oil, causing livedo reticularis, pulmonary infiltrates, and elevated levels of creatine kinase.
Scleroderma treatment
In some cases, the skin problems associated with scleroderma fade away on their own in three to five years. The type of scleroderma that affects internal organs usually worsens with time.
Medications
No drug has been developed that can stop the underlying process of scleroderma — the overproduction of collagen. But a variety of medications can help control scleroderma symptoms or help prevent complications. To accomplish this, these drugs may:
- Dilate blood vessels. Blood pressure medications that dilate blood vessels may help prevent lung and kidney problems and may help treat Raynaud’s disease.
- Suppress the immune system. Drugs that suppress the immune system (immunosuppressants), such as those taken after organ transplants, may help reduce scleroderma symptoms.
- Reduce stomach acid. Medications such as omeprazole (Prilosec) can relieve symptoms of acid reflux.
- Prevent infections. Antibiotic ointment, cleaning and protection from the cold may help prevent infection of fingertip ulcers caused by Raynaud’s disease.
- Regular influenza and pneumonia vaccinations can help protect lungs that have been damaged by scleroderma.
- Relieve pain. If over-the-counter pain relievers don’t help enough, you can ask your doctor to prescribe stronger medications.
Physical therapy
Physical or occupational therapists can help you to:
- Manage pain
- Improve your strength and mobility
- Maintain independence with daily tasks
Surgery
Used as a last resort, surgical options for scleroderma complications may include:
- Amputation. If finger ulcers caused by severe Raynaud’s disease have developed gangrene, amputation may be necessary.
- Lung transplants. People who have developed high blood pressure in the arteries to their lungs (pulmonary hypertension) may be candidates for lung transplants.
Systemic scleroderma treatment
Treatments help with symptoms and may modify the disease outcome, especially early in the disease course. They focus on suppressing inflammation and dilating abnormal / constricted blood vessels. Some newer treatments target specific immunological pathways and signalling molecules.
General advice
- It is absolutely essential for smokers to stop smoking.
- Avoid vasoconstrictive drugs, such as decongestants, amphetamines, ergotamine.
Fatigue, weakness and generalized musculoskeletal symptoms can be debilitating.
- Hydroxychloroquine may help these constitutional symptoms.
- Gentle controlled exercise may also be of benefit.
- Simple or more complex analgesia may be required.
Anti-fibrotic therapies
Topical therapies
- Warm wax baths (hands)
- Tacrolimus ointment
- Potent corticosteroids
- Calcipotriol
Physical therapies
- Phototherapy, especially UVA1 or PUVA
Systemic therapies
- Mycophenolate mofetil
- Methotrexate
- Cyclophosphamide
- Corticosteroids (with caution)
- Rituximab (B-cell depletion therapy)
- Intravenous immunoglobulin
- New targeted therapies
- Anti-interleukin 6 (tocilizumab)
- Anti-cytotoxic T-cell ligand inhibitor (abatacept)
- Tyrosine kinase inhibitors (nintedanib)
- Transforming growth factor β inhibitors (fresolimumab, perfenidone)
- Anti-Interleukin 4 / 13 drugs
- Autologous stem cell transplant
Vasodilation
- Endothelin 1 antagonists (bosentan)
- Phosphodiesterase-5 inhibitors (tadalifil)
- Guanylate cyclase agonists (riociguat)
- Prostacyclin agonists (epoprostenol; selexipag)
Treatment of skin manifestations
Raynaud phenomenon
- Avoid triggers (smoking cessation, protect from cold)
- General measures; double lined gloves, exothermic hand / feet warmers
- Natural therapies (unproven)
- Vitamin C, Vitamin E
- Gamolenic acid
- Medical therapies
- Fluoxetine
- Losartan
- Diltiazem, nifedipine
- Sildenafil, tadalafil
- GTN patches
- Iloprost
- Botulinum toxin
Digital ulcers
- Ulcer prevention
- Emollients
- Avoid smoking, cold (double lined gloves), trauma
- Ulcer treatment
- Localized dressings; protect and keep moist areas of impending ulceration and ulcerated areas
- Systemic vasodilators
- Surgery
- Amputation
- Botulinum toxin injections
- Digital sympathectomy
- Prevention of ulcer complications
- Pain management
- Infection – 10% infection rate per year
Telangiectases
- Cosmetic camouflage (green tinted makeup)
- Vascular laser or intense pulsed light therapy
Calcinosis
Calcinosis is difficult to manage and there is poor evidence for therapies listed below:
- Medical therapies
- Tetracycline antibiotics (6–12 week courses)
- Diltiazem
- Bisphosphonates
- Sodium thiosulfate (systemic and localised injections are reported)
- Cinacalcet
- Physical interventions
- Surgical excision
- Curettage and cautery
- Dental drill removal
- Ablative laser
- Extracorporeal shock wave lithotripsy
Pruritus
- Emollient, soap substitutes
- Topical corticosteroids
- Antihistamines
- Montelukast
- Systemic glucocorticoids (with caution)
Facial fat loss
- Autologous fat transfer
Treatment of internal organ manifestations
Interstitial lung disease
- Antifibrotic, immunosuppressive therapies (see above)
Pulmonary artery hypertension
- Vasodilatory therapies (see above)
Renal disease
- Monitor blood pressure regularly
- Avoid corticosteroids (especially if RNAPIII +ve)
- Angiotensin converting enzyme inhibitors
Gastrointestinal tract
- Upper GIT: gastro-oesophageal reflux, dyspepsia, dysphagia, aspiration
- Protein pump inhibitors, such as oomeprazole, ranitidine
- Treat Helicobacter pylori
- Prokinetics: domperidone, erythromycin, metoclopramide
- N-acetylcysteine
- Lower GIT: malabsorption, bacterial overgrowth, diarrhoea, constipation, bloating, pain, anorectal dysfunction
- Nutritional supplements, advice from dietician
- Creon
- Probiotics
- Cyclical antibiotics (for example, ciprofloxacin for 10 days)
- Stool softeners
Home remedies
You can take an active part in treating your scleroderma. Be sure to take your medications as prescribed, keep your physical therapy appointments, and call your doctor if you notice new symptoms.
You can take a number of steps to help manage your symptoms of scleroderma:
- Stay active. Exercise keeps your body flexible, improves circulation and relieves stiffness. Range-of-motion exercises can help keep your skin and joints flexible.
- Don’t smoke. Nicotine causes blood vessels to contract, making Raynaud’s disease worse. Smoking can also cause permanent narrowing of your blood vessels. Quitting smoking is difficult — ask your doctor for help.
- Manage heartburn. Avoid foods that give you heartburn or gas. Also avoid late-night meals. Elevate the head of your bed to keep stomach acid from backing up into your esophagus (reflux) as you sleep. Antacids may help relieve symptoms.
- Protect yourself from the cold. Wear warm mittens for protection anytime your hands are exposed to cold — even when you reach into a freezer. When you’re outside in the cold, cover your face and head and wear layers of warm clothing.
Here are some ways to take care of your skin:
- Skin problems. With scleroderma, collagen builds up in the skin. Too much of it can make your skin dry and stiff. To help, you can:
- Use oil-based creams and lotions after every bath.
- Use sunscreen.
- Use a humidifier at home.
- Avoid hot baths or showers.
- Avoid strong soaps, cleaners, and chemicals. Wear rubber gloves if you have to use those products.
- Exercise regularly.
- Cosmetic problems. Scleroderma can damage your skin and change how it looks. These skin changes can affect your self-image. Ways to fix skin damage include:
- Lasers that take away red spots on the hands and face.
- Plastic surgery in areas where the disease is not active.
If you have systemic scleroderma, the disease may affect other parts of your body, besides just your skin. Here are some common treatments and things to watch for if you have systemic scleroderma:
- Raynaud’s phenomenon. Most people with scleroderma have Raynaud’s phenomenon. It can affect the fingers, feet, and hands. It makes them change color if you are too cold or anxious. To help, you can:
- Not smoke.
- Dress warm, and keep hands and feet warm.
- Do exercises that relax the body.
- Ask about medicines that open small blood vessels and help with blood flow.
- Ask about medicines that treat skin sores and ulcers.
- Stiff, painful joints. Stiffness and pain come from hard skin around joints and joint swelling. To help, you can:
- Do stretching exercises that help with joint motion.
- Exercise regularly (swimming is best).
- Take medicine to help ease pain or swelling. Ask your doctor which are the best for you to take.
- Learn to do daily tasks in ways that put less stress on the joints.
- Dry mouth and dental problems. If you have tight skin on your face, you may have trouble caring for your teeth. Dry mouth speeds up tooth decay. Harm to tissues in the mouth can loosen teeth. To avoid problems:
- Brush and floss your teeth each day.
- Have frequent dental checkups.
- See your dentist if you have mouth sores, mouth pain, or loose teeth.
- Ask your dentist about special rinses and toothpastes.
- Learn ways to keep your mouth and face flexible.
- Keep your mouth moist. You can drink lots of water or suck on ice chips. You can also chew gum or suck on hard candy that has no sugar added.
- Avoid mouthwash that has alcohol.
- If dry mouth still bothers you, ask your doctor about helpful medicines.
- Gastrointestinal problems. Digestive problems can include heartburn, trouble swallowing, feeling full as soon as you start eating, diarrhea, constipation, and gas. To help, you can:
- Eat small, frequent meals.
- Stand or sit for 1 to 3 hours after eating.
- Use blocks to raise the head of your bed.
- Avoid late-night meals, spicy or fatty foods, alcohol, and caffeine.
- Eat moist, soft foods, and chew them well.
- Ask your doctor about medicines for diarrhea, constipation, and heartburn.
- Lung damage. Lung problems with systemic scleroderma can include loss of lung function, severe lung disease, lung tissue scarring, and high blood pressure in the artery that carries blood from your heart to your lungs. Watch for signs of lung disease, such as:
- Fatigue
- Shortness of breath
- Problems with breathing
- Swollen feet.
- As soon as your skin starts to thicken, see your doctor. Get regular flu and pneumonia shots.
- Heart problems. Systemic scleroderma can sometimes cause scarring and weakness in your heart, as well as swelling of the heart muscle and a heartbeat that isn’t normal. These problems can all be treated with help from your doctor.
- Kidney problems. Scleroderma can cause very high blood pressure and kidney failure in some people. Talk to your doctor about what symptoms to look for so you can spot problems right away. You should:
- Check your blood pressure often.
- Check your blood pressure if you have new symptoms.
- Call your doctor if your blood pressure is higher than normal.
- Take the medicines your doctor prescribes.
Coping and support
As is true with other chronic diseases, living with scleroderma can place you on a roller coaster of emotions. Here are some suggestions to help you even out the ups and downs:
- Maintain normal daily activities as best you can.
- Pace yourself and be sure to get the rest that you need.
- Stay connected with friends and family.
- Continue to pursue hobbies that you enjoy and are able to do.
Keep in mind that your physical health can have a direct impact on your mental health. Denial, anger and frustration are common with chronic illnesses.
At times, you may need additional tools to deal with your emotions. Professionals, such as therapists or behavior psychologists, may be able to help you put things in perspective. They can also help you develop coping skills, including relaxation techniques.
Joining a support group (Scleroderma Foundation at http://www.scleroderma.org), where you can share experiences and feelings with other people, is often a good approach. Ask your doctor what support groups are available in your community.
Systemic scleroderma monitoring
Monitoring of progress and treatment response is vital in systemic scleroderma.
Skin
The skin is usually monitored clinically using the modified Rodnan Skin Score, which gives an indication of the extent and severity of the cutaneous scleroderma, which also reflects the severity and risk of internal organ involvement.
- A score of 0 (normal skin) to 3 (skin not able to be pinched) is assigned for skin tightness for 17 body sites.
- The cumulative score is calculated out of a total of 51; severe skin involvement is defined by a score of > 20.
- A high modified Rodnan Skin Score is an independent risk factor for a poorer overall outcome.
Interlabial distance (mouth opening) can also be measured
Microstomia is defined as an inter-labial distance of less than 4.5 cm.
Internal organs
Routine annual screening for interstitial lung disease and pulmonary artery hypertension should include:
- Pulmonary function tests
- Transthoracic echocardiogram
- ECG
The DETECT score is a screening tool for pulmonary artery hypertension that uses pulmonary function tests (FVC and DLCO), serum urate, serum NTproBNP and other features to provide a predictive score. A right heart catheter may be indicated in some.
- High resolution CT scan may be used to characterize interstitial lung disease.
- Blood pressure and renal function monitor renal disease.
- Other organ monitoring is performed on an individualized basis, and can include cardiac MRI, esophageal manometry and gastrointestinal endoscopy.
- Adigun R, Goyal A, Hariz A. Systemic Sclerosis (Scleroderma) [Updated 2024 Apr 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430875[↩][↩][↩][↩][↩][↩][↩]
- Barnett AJ, Miller MH, Littlejohn GO. A survival study of patients with scleroderma diagnosed over 30 years (1953-1983): the value of a simple cutaneous classification in the early stages of the disease. J Rheumatol. 1988 Feb;15(2):276-83.[↩]
- Mayes MD, Lacey JV Jr, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, Schottenfeld D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003 Aug;48(8):2246-55. doi: 10.1002/art.11073[↩]
- Elhai M, Meune C, Avouac J, Kahan A, Allanore Y. Trends in mortality in patients with systemic sclerosis over 40 years: a systematic review and meta-analysis of cohort studies. Rheumatology (Oxford). 2012 Jun;51(6):1017-26. doi: 10.1093/rheumatology/ker269[↩]
- Nikpour M, Baron M. Mortality in systemic sclerosis: lessons learned from population-based and observational cohort studies. Curr Opin Rheumatol. 2014 Mar;26(2):131-7. doi: 10.1097/BOR.0000000000000027[↩]
- Nihtyanova SI, Tang EC, Coghlan JG, Wells AU, Black CM, Denton CP. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: a retrospective cohort study. QJM. 2010 Feb;103(2):109-15. doi: 10.1093/qjmed/hcp174[↩]
- Cottrell TR, Wise RA, Wigley FM, Boin F. The degree of skin involvement identifies distinct lung disease outcomes and survival in systemic sclerosis. Ann Rheum Dis. 2014 Jun;73(6):1060-6. doi: 10.1136/annrheumdis-2012-202849[↩]
- Winstone TA, Assayag D, Wilcox PG, Dunne JV, Hague CJ, Leipsic J, Collard HR, Ryerson CJ. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: a systematic review. Chest. 2014 Aug;146(2):422-436. doi: 10.1378/chest.13-2626[↩]
- Chung L, Domsic RT, Lingala B, Alkassab F, Bolster M, Csuka ME, Derk C, Fischer A, Frech T, Furst DE, Gomberg-Maitland M, Hinchcliff M, Hsu V, Hummers LK, Khanna D, Medsger TA Jr, Molitor JA, Preston IR, Schiopu E, Shapiro L, Silver R, Simms R, Varga J, Gordon JK, Steen VD. Survival and predictors of mortality in systemic sclerosis-associated pulmonary arterial hypertension: outcomes from the pulmonary hypertension assessment and recognition of outcomes in scleroderma registry. Arthritis Care Res (Hoboken). 2014 Mar;66(3):489-95. doi: 10.1002/acr.22121[↩]
- Scleroderma. https://emedicine.medscape.com/article/331864-overview#a6[↩][↩][↩]
- Hissaria P, Roberts-Thomson PJ, Lester S, Ahern MJ, Smith MD, Walker JG. Cigarette smoking in patients with systemic sclerosis reduces overall survival: comment on the article by Hudson et al. Arthritis Rheum. 2011 Jun;63(6):1758-9. doi: 10.1002/art.30352[↩]
- Pokeerbux MR, Giovannelli J, Dauchet L, Mouthon L, Agard C, Lega JC, Allanore Y, Jego P, Bienvenu B, Berthier S, Mekinian A, Hachulla E, Launay D. Survival and prognosis factors in systemic sclerosis: data of a French multicenter cohort, systematic review, and meta-analysis of the literature. Arthritis Res Ther. 2019 Apr 3;21(1):86. doi: 10.1186/s13075-019-1867-1[↩]
- Systemic Sclerosis. https://emedicine.medscape.com/article/1066280-overview#a2[↩][↩]
- Alba MA, Velasco C, Simeón CP, Fonollosa V, Trapiella L, Egurbide MV, Sáez L, Castillo MJ, Callejas JL, Camps MT, Tolosa C, Ríos JJ, Freire M, Vargas JA, Espinosa G; RESCLE Registry. Early- versus late-onset systemic sclerosis: differences in clinical presentation and outcome in 1037 patients. Medicine (Baltimore). 2014 Mar;93(2):73-81. doi: 10.1097/MD.0000000000000018[↩]
- Woodworth TG, Suliman YA, Li W, Furst DE, Clements P. Scleroderma renal crisis and renal involvement in systemic sclerosis. Nat Rev Nephrol. 2016 Nov;12(11):678-691. doi: 10.1038/nrneph.2016.124. Epub 2016 Sep 19. Erratum in: Nat Rev Nephrol. 2018 Feb;14(2):137. doi: 10.1038/nrneph.2017.183[↩]
- Jancin B. Be vigilant for scleroderma renal crisis. Rheumatology News. https://www.mdedge.com/rheumatology/article/223925/lupus-connective-tissue-diseases/be-vigilant-scleroderma-renal-crisis[↩]
- Heřmánková B, Špiritović M, Šmucrová H, Oreská S, Štorkánová H, Komarc M, Pavelka K, Šenolt L, Vencovský J, Bečvář R, Tomčík M. Female Sexual Dysfunction and Pelvic Floor Muscle Function Associated with Systemic Sclerosis: A Cross-Sectional Study. Int J Environ Res Public Health. 2022 Jan 5;19(1):612. doi: 10.3390/ijerph19010612[↩][↩][↩]
- Frerix M, Kröger K, Szalay G, Müller-Ladner U, Tarner IH. Is osteonecrosis of the lunate bone an underestimated feature of systemic sclerosis? A case series of nine patients and review of literature. Semin Arthritis Rheum. 2016 Feb;45(4):446-54. doi: 10.1016/j.semarthrit.2015.07.010[↩]
- Scleroderma. https://emedicine.medscape.com/article/331864-overview#a4[↩][↩]
- Dospinescu P, Jones GT, Basu N. Environmental risk factors in systemic sclerosis. Curr Opin Rheumatol. 2013 Mar;25(2):179-83. doi: 10.1097/BOR.0b013e32835cfc2d[↩]
- Nietert PJ, Sutherland SE, Silver RM, Pandey JP, Knapp RG, Hoel DG, Dosemeci M. Is occupational organic solvent exposure a risk factor for scleroderma? Arthritis Rheum. 1998 Jun;41(6):1111-8. doi: 10.1002/1529-0131(199806)41:6<1111::AID-ART19>3.0.CO;2-J. Erratum in: Arthritis Rheum 1998 Aug;41(8):1512.[↩]
- Marie I, Gehanno JF, Bubenheim M, Duval-Modeste AB, Joly P, Dominique S, Bravard P, Noël D, Cailleux AF, Weber J, Lagoutte P, Benichou J, Levesque H. Prospective study to evaluate the association between systemic sclerosis and occupational exposure and review of the literature. Autoimmun Rev. 2014 Feb;13(2):151-6. doi: 10.1016/j.autrev.2013.10.002[↩]
- Rubio-Rivas M, Moreno R, Corbella X. Occupational and environmental scleroderma. Systematic review and meta-analysis. Clin Rheumatol. 2017 Mar;36(3):569-582. doi: 10.1007/s10067-016-3533-1[↩]
- Barragán-Martínez C, Speck-Hernández CA, Montoya-Ortiz G, Mantilla RD, Anaya JM, Rojas-Villarraga A. Organic solvents as risk factor for autoimmune diseases: a systematic review and meta-analysis. PLoS One. 2012;7(12):e51506. doi: 10.1371/journal.pone.0051506[↩]
- McCormic ZD, Khuder SS, Aryal BK, Ames AL, Khuder SA. Occupational silica exposure as a risk factor for scleroderma: a meta-analysis. Int Arch Occup Environ Health. 2010 Oct;83(7):763-9. doi: 10.1007/s00420-009-0505-7[↩][↩]
- Barbosa NS, Wetter DA, Wieland CN, Shenoy NK, Markovic SN, Thanarajasingam U. Scleroderma Induced by Pembrolizumab: A Case Series. Mayo Clin Proc. 2017 Jul;92(7):1158-1163. doi: 10.1016/j.mayocp.2017.03.016[↩]
- Moroncini G, Mori S, Tonnini C, Gabrielli A. Role of viral infections in the etiopathogenesis of systemic sclerosis. Clin Exp Rheumatol. 2013 Mar-Apr;31(2 Suppl 76):3-7. https://www.clinexprheumatol.org/abstract.asp?a=7306[↩]
- Hertzman PA, Blevins WL, Mayer J, Greenfield B, Ting M, Gleich GJ. Association of the eosinophilia-myalgia syndrome with the ingestion of tryptophan. N Engl J Med. 1990 Mar 29;322(13):869-73. doi: 10.1056/NEJM199003293221301[↩]
- Arnett FC, Cho M, Chatterjee S, Aguilar MB, Reveille JD, Mayes MD. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001 Jun;44(6):1359-62. doi: 10.1002/1529-0131(200106)44:6<1359::AID-ART228>3.0.CO;2-S[↩]
- Assassi S, Arnett FC, Reveille JD, Gourh P, Mayes MD. Clinical, immunologic, and genetic features of familial systemic sclerosis. Arthritis Rheum. 2007 Jun;56(6):2031-7. doi: 10.1002/art.22647[↩]
- González-Serna D, Shi C, Kerick M, Hankinson J, Ding J, McGovern A, Tutino M, Villanueva-Martin G, Ortego-Centeno N, Callejas JL, Martin J, Orozco G. Identification of Mechanisms by Which Genetic Susceptibility Loci Influence Systemic Sclerosis Risk Using Functional Genomics in Primary T Cells and Monocytes. Arthritis Rheumatol. 2023 Jun;75(6):1007-1020. doi: 10.1002/art.42396[↩]
- Almaabdi K, Ahmad Z, Johnson SR. Advanced Autoantibody Testing in Systemic Sclerosis. Diagnostics (Basel). 2023 Feb 23;13(5):851. doi: 10.3390/diagnostics13050851[↩]
- Zanatta E, Huscher D, Ortolan A, Avouac J, Airò P, Balbir-Gurman A, Siegert E, Matucci Cerinic M, Cozzi F, Riemekasten G, Hoffmann-Vold AM, Distler O, Gabrielli A, Heitmann S, Hunzelmann N, Montecucco C, Morovic-Vergles J, Ribi C, Doria A, Allanore Y; EUSTAR collaborators. Phenotype of limited cutaneous systemic sclerosis patients with positive anti-topoisomerase I antibodies: data from the EUSTAR cohort. Rheumatology (Oxford). 2022 Nov 28;61(12):4786-4796. doi: 10.1093/rheumatology/keac188[↩]
- Steen V, Domsic RT, Lucas M, Fertig N, Medsger TA Jr. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum. 2012 Sep;64(9):2986-94. doi: 10.1002/art.34482[↩]
- Alvarenga Fernandes D, Garcez Teixeira CE, Toledo Del Rio AP, Sachetto Z, Reis F. Intestinal pneumatosis as a manifestation of systemic sclerosis. Rev Esp Enferm Dig. 2023 Apr;115(4):220-221. doi: 10.17235/reed.2023.9428/2022[↩]
- Adarsh MB, Sharma SK, Prasad KK, Dhir V, Singh S, Sinha SK. Esophageal manometry, esophagogastroduodenoscopy, and duodenal mucosal histopathology in systemic sclerosis. JGH Open. 2019 Mar 20;3(3):206-209. doi: 10.1002/jgh3.12138[↩]
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013 Nov;65(11):2737-47. doi: 10.1002/art.38098[↩]
- Lamback EB, Resende FS, Lenzi TC. Eosinophilic fasciitis. An Bras Dermatol. 2016 Sep-Oct;91(5 suppl 1):57-59. doi: 10.1590/abd1806-4841.20164683[↩][↩]
- Niklas K, Niklas A, Puszczewicz M. Eozynofilowe zapalenie powięzi [Eosinophilic fasciitis]. Postepy Hig Med Dosw (Online). 2015 Jan 2;69:488-95. Polish. doi: 10.5604/17322693.1149872[↩]
- Kim S, Park TH, Lee SM, Kim YH, Cho MK, Whang KU, Kim HS. Scleromyxedema with multiple systemic involvement: Successful treatment with intravenous immunoglobulin. Dermatol Ther. 2020 May;33(3):e13378. doi: 10.1111/dth.13378[↩][↩]
- Gambichler T, Susok L, Doerler M, Dickel H, Chatzipantazi M. Löfgren syndrome associated with scleroedema adultorum of Buschke. Clin Exp Dermatol. 2023 Jan 20;48(1):39-40. doi: 10.1093/ced/llac021[↩][↩]





{kind=link}