Contents
Leigh syndrome
Leigh syndrome also called Leigh’s disease, classical Leigh syndrome, subacute necrotizing encephalopathy or subacute necrotizing encephalomyelopathy (SNEM) is a inherited severe progressive neurodegenerative mitochondrial disease that affects the central nervous system (brain and spinal cord), leading to a progressive degeneration of brain tissue that is characterized by progressive loss of mental and movement abilities (psychomotor regression) and resulting in early death within two to three years, often due to respiratory failure 1, 2, 3, 4, 5, 6. Leigh syndrome is the most common infantile form of mitochondrial disease, usually becomes apparent in infants and young children of 3 months to 2 years of age and affecting around 1 in 40,000 individuals 3, 7, 6. Moreover, the precise prevalence of Leigh syndrome is not known as many cases are wrongly diagnosed 8. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly.
In 1951, a British psychiatrist and neuropathologist Denis Archibald Leigh through postmortem investigation of a 7-month-old boy with progressive neurologic symptoms gave a very clear definition of subacute necrotizing encephalomyelopathy (SNEM), where he described a neuropathological entity characterized by early-onset development of death of tissues (necrotizing lesions) in the grey nuclei of the brainstem, subthalamic region, and basal ganglia, including often the deep structures of the cerebellum and descending up to the rostral segment of the spinal cord 9, 10. These lesions are usually symmetric, with neuronal loss, initial inflammation followed by gliosis, and maintenance of capillary hyperproliferation 9, 10. Any Leigh syndrome or Leigh-like syndrome (whose definition is much more uncertain) must contain these essential neuropathological features 11. Since the first description of Leigh syndrome based on histopathological findings, many tests have tried to support the diagnosis revealing a damage in mitochondrial metabolism 12.
Leigh syndrome is defined in the Online Mendelian Inheritance in Man Database as follows: (1) A neurodegenerative disease with variable symptoms (2) Caused by mitochondrial dysfunction from a hereditary genetic defect, and (3) Accompanied by bilateral central nervous system (CNS) lesions 13, 14. However, these criteria are not always met, and, in those cases, the term “Leigh-like syndrome” is preferred 15, 16. The absence of well-defined criteria for Leigh syndrome results from the broadest genetic heterogeneity; even non-mitochondrial disorders also may present as Leigh syndrome 17. Leigh syndrome often shows a progressive decline of central nervous system (brain and spinal cord) because of focal necrotizing lesions of the basal ganglia, cerebellum, diencephalon, or brainstem, as already mentioned 18. Leigh syndrome and Leigh-like syndromes have defects in the respiratory chain, coenzyme Q, or pyruvate dehydrogenase complex 19.
The first signs of Leigh syndrome seen in infancy are usually vomiting, diarrhea, and difficulty swallowing (dysphagia), which disrupts eating 6. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive). Severe muscle and movement problems are common in Leigh syndrome. Affected individuals may develop weak muscle tone (hypotonia), involuntary muscle contractions (dystonia), and problems with movement and balance (ataxia). Loss of sensation and weakness in the limbs (peripheral neuropathy), common in people with Leigh syndrome, may also make movement difficult 6. According to a meta-analysis by Chang et al. 20, the most common clinical features of Leigh syndrome were developmental delay, hypotonia, respiratory dysfunction, epilepsy, reduced feeding, and weakness.
Several other features may occur in people with Leigh syndrome. Many individuals with Leigh syndrome develop weakness or paralysis of the muscles that move the eyes (ophthalmoplegia); rapid, involuntary eye movements (nystagmus); strabismus or “lazy eyes” (a condition where the eyes do not point in the same direction with one or both eyes either turning inward [esotropia], outward [exotropia], upward [hypertropia], or downward [hypotropia]); or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy) and pigmentary retinopathy 20, 21, 6. Severe breathing problems are common, and these problems can worsen until they cause acute respiratory failure 6. Some affected individuals also develop heart abnormalities such as hypertrophic cardiomyopathy (a thickening of the heart muscle that forces the heart to work harder to pump blood) or dilated cardiomyopathy (a heart muscle disease where the heart’s main pumping chamber, left ventricle, enlarges and weakens, making it difficult for the heart to pump blood efficiently) and conduction defects such as Wolff–Parkinson–White syndrome 22, 23, 6. In addition, a substance called lactate can build up in the body, and excessive amounts are often found in the blood, urine, or the fluid that surrounds and protects the brain and spinal cord (cerebrospinal fluid [CSF]) of people with Leigh syndrome 6.
The signs and symptoms of Leigh syndrome are caused in part by patches of damaged tissue (lesions) that develop in the brains of people with Leigh syndrome. A medical procedure called magnetic resonance imaging (MRI) reveals characteristic lesions in certain regions of the brain. These regions include the basal ganglia, which help control movement; the cerebellum, which controls the ability to balance and coordinates movement; and the brainstem, which connects the brain to the spinal cord and controls functions such as swallowing and breathing. The brain lesions are often accompanied by loss of the myelin coating around nerves (demyelination), which reduces the ability of the nerves to activate muscles used for movement or relay sensory information from the rest of the body back to the brain.
Leigh syndrome has been reported to be caused by defects of 16 mitochondrial DNA (mtDNA) genes encoding 10 oxidative phosphorylation (OXPHOS) subunits and 6 transfer RNAs (tRNAs) and nuclear DNA (nDNA) defects in more than 100 different genes, most commonly by the SURF1 mutation 8, 24, 25. In humans, most genes are found in DNA in the cell’s nucleus, called nuclear DNA (nDNA). However, some genes are found in DNA in specialized structures in the cell called mitochondria. This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a mutation in nuclear DNA (nDNA), about 20 percent to 47 percent of cases have multiple mitochondrial DNA (mtDNA) deletions 22, 26.
Leigh syndrome caused by mitochondrial DNA (mtDNA) mutations can be maternally inherited or sporadic. Maternally inherited cases usually happen in a clinically unaffected mother. Inheritance of nuclear-encoded (nDNA) Leigh syndrome is typically autosomal recessive and rarely X-linked 8.
Most genes associated with Leigh syndrome are involved in the process of energy production in mitochondria. Mitochondria use oxygen to convert the energy from food into a form cells can use through a process called oxidative phosphorylation. Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell’s main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene variants associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These variants reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
Disruption of complex I, also called NADH:ubiquinone oxidoreductase, is the most common cause of Leigh syndrome, accounting for nearly one third of cases of Leigh syndrome 6. At least 25 genes involved in the formation of complex I, found in either nuclear or mitochondrial DNA, have been associated with Leigh syndrome.
Disruption of complex IV, also called cytochrome C oxidase (COX), is also a common cause of Leigh syndrome, underlying approximately 15 percent of cases. One of the most frequently altered genes in Leigh syndrome is SURF1. The SURF1 gene is found in nuclear DNA (nDNA) provides instructions for making a protein that helps assemble the cytochrome C oxidase (COX) protein complex (complex IV). This complex, which is involved in the last step of electron transfer in oxidative phosphorylation, provides the energy that will be used in the next step of the process to generate ATP (adenosine triphosphate). Mutations in the SURF1 gene typically lead to an abnormally short SURF1 protein that is broken down in cells, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal cytochrome C oxidase (COX) complexes, which impairs mitochondrial energy production.
The most common mitochondrial DNA (mtDNA) mutation in Leigh syndrome affects the MT-ATP6 gene, which provides instructions for making a piece of complex V, also known as the ATP synthase protein complex. Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene mutations, found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mitochondrial DNA (mtDNA) mutations associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
Other gene mutations associated with Leigh syndrome decrease the activity of one or more oxidative phosphorylation protein complexes or affect additional steps related to energy production. For example, Leigh syndrome can be caused by variants in genes that form the pyruvate dehydrogenase complex or coenzyme Q10, both of which are involved in mitochondrial energy production. Mutations in genes that direct the replication of mtDNA or the production of mitochondrial proteins can also disrupt mitochondrial energy production.
Although the exact mechanism is unclear, researchers believe that impaired oxidative phosphorylation can lead to cell death because of decreased energy available in the cell. Certain tissues that require large amounts of energy, such as the brain, muscles, and heart, seem especially sensitive to decreases in cellular energy. Cell death in the brain likely causes the characteristic lesions seen in Leigh syndrome, which contribute to the signs and symptoms of Leigh syndrome. Cell death in other sensitive tissues may also contribute to the features of Leigh syndrome.
Consensus on Leigh syndrome diagnosis is yet to be determined; however, Leigh syndrome is suspected through the hallmarks of the disease along with findings suggestive of brainstem dysfunction in addition to T2 weighted brain MRI lesions and accessory laboratory findings 20. Brain MRI findings typically show bilateral symmetrical supra-tentorial (basal ganglia, thalamus, and sub-thalamus) and/or infra-tentorial (brainstem and dentate nuclei) lesions. A study by Ardissone et al 27 presented a predominating basal ganglia involvement of 90.2%. They also showed that both supra and infra-tentorial involvement is dominant in cases of both mtDNA (74%) and -nDNA (67%) variants, while isolated infra-tentorial variants are rare 27. Extensive research is being conducted to find genetic correlations with MRI findings of Leigh syndrome. For example, a retrospective cohort found significant associations between the SURF1 variant and inferior olivary nuclei lesions 28.
Abnormal laboratory findings may yield elevated blood, urine, and CSF lactate levels. Additional deficiencies may be observed in respiratory chain complexes through enzyme assays and pyruvate dehydrogenase complex 29. However, these laboratory findings are not consistently present. Therefore, confirmatory tests with genetic testings are required for a definitive diagnosis and the identification of specific gene mutations of Leigh syndrome 30.
There is no disease-modifying therapy available for Leigh syndrome 31, 32. Current therapies include supplements coenzyme Q10 (CoQ10) and its derivatives, thiamine (vitamin B1), riboflavin (vitamin B2) and vitamin C, pyruvate, dichloroacetate, and a ketogenic diet 33, 34, 32.
Figure 1. Leigh syndrome
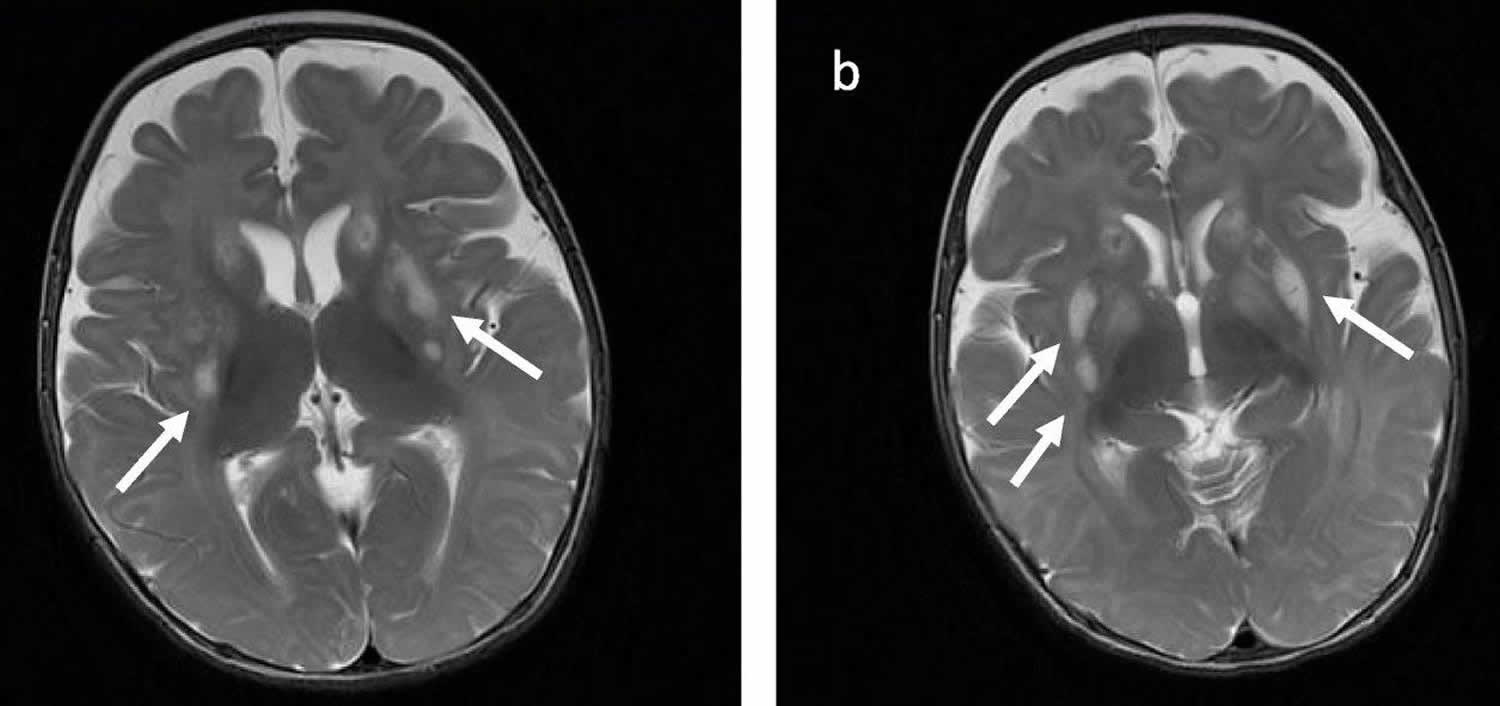
Footnotes: Brain MRI images in axial T2-weighted acquisition (a, b) demonstrating abnormal sign in bilateral basal ganglia (white arrows), which is a common finding of Leigh Syndrome.
[Source 2 ]What is mitochondria?
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called adenosine triphosphate (ATP) that your cells can use 35. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP (adenosine triphosphate). Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus.
The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. The parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 36. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 37. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 2. Mitochondria cell
What is mitochondrial disease?
Mitochondrial diseases can affect almost any part of your body, including the cells of your:
- Brain – developmental delays, dementia, migraines, autistic features, seizure, stroke, atypical cerebral palsy, learning disabilities, problems with coordination and balance (ataxia).
- Nerves – neuropathy (nerve damage), fainting, zero reflexes, heat/cold intolerance, pain.
- Muscles – weakness, cramping, reflux, vomiting, constipation, diarrhea, low muscle tone (hypotonia), dysmotility
- Kidneys – renal tube failure
- Heart – heart defects, heart blocks, cardiomyopathy.
- Liver – low blood sugar, liver failure.
- Eyes – vision loss, ptosis, optic atrophy, strabismus, ophthalmoplegia, retinitis pigmentosa.
- Ears – hearing loss
- Pancreas – diabetes, pancreatic failure, parathyroid failure.
Figure 3. Mitochondrial diseases
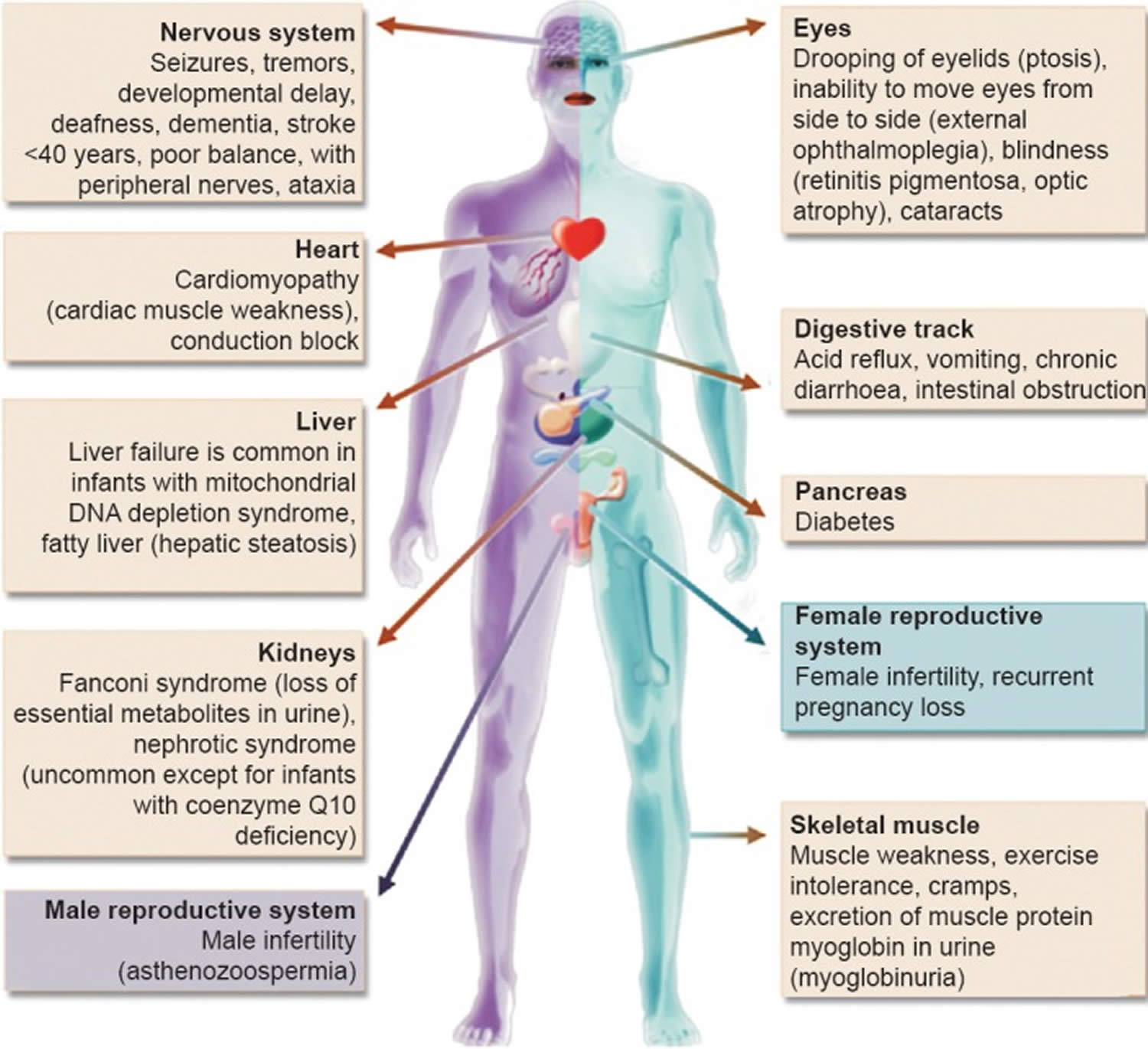
Footnote: Clinical features and the organs affected by mitochondrial diseases.
[Source 38 ]Figure 4. Mitochondrial diseases signs and symptoms
Are mitochondrial diseases difficult to diagnose?
Yes. Because mitochondrial diseases affect so many different organs and tissues of your body, and you may have many different symptoms, mitochondrial diseases can be difficult to diagnose. There’s no single laboratory test that can diagnose a mitochondrial disease. This is why a referral to a medical facility with physicians who specialize in these diseases is critical to making the diagnosis.
Leigh syndrome cause
Leigh syndrome has been reported to be caused by defects of 16 mitochondrial DNA (mtDNA) genes encoding 10 oxidative phosphorylation (OXPHOS) subunits and 6 transfer RNAs (tRNAs) and nuclear DNA (nDNA) defects in more than 100 different genes, most commonly by the SURF1 mutation 8, 24, 25. In humans, most genes are found in DNA in the cell’s nucleus, called nuclear DNA (nDNA). However, some genes are found in DNA in specialized structures in the cell called mitochondria. This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a mutation in nuclear DNA (nDNA), about 20 percent to 47 percent of cases have multiple mitochondrial DNA (mtDNA) deletions 22, 26.
Leigh syndrome caused by mitochondrial DNA (mtDNA) mutations can be maternally inherited or sporadic. Maternally inherited cases usually happen in a clinically unaffected mother. Inheritance of nuclear-encoded (nDNA) Leigh syndrome is typically autosomal recessive and rarely X-linked 8.
Most genes associated with Leigh syndrome are involved in the process of energy production in mitochondria. Mitochondria use oxygen to convert the energy from food into a form cells can use through a process called oxidative phosphorylation. Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell’s main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene variants associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These variants reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
Disruption of complex I, also called NADH:ubiquinone oxidoreductase, is the most common cause of Leigh syndrome, accounting for nearly one third of cases of Leigh syndrome 6. At least 25 genes involved in the formation of complex I, found in either nuclear or mitochondrial DNA, have been associated with Leigh syndrome.
Disruption of complex IV, also called cytochrome C oxidase (COX), is also a common cause of Leigh syndrome, underlying approximately 15 percent of cases. One of the most frequently altered genes in Leigh syndrome is SURF1. The SURF1 gene is found in nuclear DNA (nDNA) provides instructions for making a protein that helps assemble the cytochrome C oxidase (COX) protein complex (complex IV). This complex, which is involved in the last step of electron transfer in oxidative phosphorylation, provides the energy that will be used in the next step of the process to generate ATP (adenosine triphosphate). Mutations in the SURF1 gene typically lead to an abnormally short SURF1 protein that is broken down in cells, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal cytochrome C oxidase (COX) complexes, which impairs mitochondrial energy production.
The most common mitochondrial DNA (mtDNA) mutation in Leigh syndrome affects the MT-ATP6 gene, which provides instructions for making a piece of complex V, also known as the ATP synthase protein complex. Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene mutations, found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mitochondrial DNA (mtDNA) mutations associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
Other gene mutations associated with Leigh syndrome decrease the activity of one or more oxidative phosphorylation protein complexes or affect additional steps related to energy production. For example, Leigh syndrome can be caused by variants in genes that form the pyruvate dehydrogenase complex or coenzyme Q10, both of which are involved in mitochondrial energy production. Mutations in genes that direct the replication of mtDNA or the production of mitochondrial proteins can also disrupt mitochondrial energy production.
Although the exact mechanism is unclear, researchers believe that impaired oxidative phosphorylation can lead to cell death because of decreased energy available in the cell. Certain tissues that require large amounts of energy, such as the brain, muscles, and heart, seem especially sensitive to decreases in cellular energy. Cell death in the brain likely causes the characteristic lesions seen in Leigh syndrome, which contribute to the signs and symptoms of Leigh syndrome. Cell death in other sensitive tissues may also contribute to the features of Leigh syndrome.
Complex 1 Deficiency
Complex 1 (C1) also called NADH:ubiquinone oxidoreductase is the primary and largest complex, weighing approximately 1 MDa, whose main function involves the transfer of electrons from NADH to coenzyme Q10 while also transporting H+ ions 1. Complex 1 consists of 44 subunits, including 14 core subunits crucial for its catalytic function, with seven subunits encoded by mitochondrial DNA (mtDNA) and the remaining core subunits encoded by nuclear DNA (nDNA). Mutations in 24 Complex 1 subunit-encoding genes and several assembly factors have been linked to Leigh syndrome, highlighting the importance of Complex 1 in mitochondrial function. Notably, mutations in the nuclear DNA (nDNA)-encoded NDUFS4 gene, encoding subunit four of Complex 1, induce ’mitochondrial complex 1 deficiency, nuclear type 1′ (MC1DN1), and Leigh syndrome in pediatric patients 39. The lack of NDUFS4 in various mouse tissues leads to reduced activity and stability of complex 1. This instability causes a greater disconnect between electron influx from the NADH dehydrogenase module and the whole complex 39, 40, 41. A dystonic later-onset form of NDUFS4 was described with an onset of toes walking, dysarthric speech, nystagmus, and mental deterioration 42. NADH ubiquinone oxidoreductase core subunit S8 (NDUFS8) is another crucial core component of the iron sulfide (FeS) fragment in mitochondrial complex 1, playing a direct role in electron transfer and energy metabolism. Pathogenic variants of NDUFS8 are associated with Leigh syndrome, as well as cancer and diabetes mellitus 42, 43. Several mitochondrial DNA (mtDNA) mutations associated with Leigh syndrome are concentrated in MTND ‘hotspots’, particularly in MTND1, MTND3, and MTND5 44. Isolated complex I deficiency is the most common oxidative phosphorylation enzyme defect, leading to a diverse clinical presentation that includes Leigh syndrome. The m.10191T>C mutation in the MTND3 subunit is relatively frequent and can be either maternally inherited or a recurrent sporadic mutation. The m.10191T>C mutation in the MTND3 subunit should be considered when evaluating individuals with complex I deficiency and Leigh syndrome features 45. Defects of complex I are usually associated with visual disturbance, nystagmus, optic atrophy, and epilepsy 8.
Complex 2–5 Deficiency
Complex II subunits are all nuclear-encoded: four structural subunits (SDHA, SDHB, SDHC, and SDHD) and two known assembly factor genes (SDHAF1 and SDHAF2). Complex II is distinct in that it serves as a component of both the respiratory chain and the Krebs cycle 46. Mitochondrial diseases linked to isolated complex II deficiency are uncommon; they can result in Leigh syndrome or familial pheochromocytomas and paragangliomas 47. Interestingly, in some cases of complex II deficiency, magnetic resonance spectroscopy (MRS) of the brain can reveal a succinate peak 48.
Complex III defect involves proteins like BCS1L, TTC19, and in mouse models, PARL. Mutations in TTC19 have been found to impair the function of Complex III, leading to a diverse clinical presentation that includes Leigh syndrome 49. The clinical presentation of TTC19 disease is variable and may consist of spinocerebellar ataxia and psychiatric manifestations, with the observation that hypertrophic olivary degeneration in the brain MRI may be of diagnostic value 50. PARL deficiency, as shown in mouse models, leads to forms of Leigh-like syndrome51. A single variant in one subunit (UQCRQ) has been reported in more than 20 affected individuals of a consanguineous Israeli family 52.
Complex IV comprises three mtDNA subunits (COX I to III encoded by MT-CO1 to 3) and three nuclear-encoded subunits (COX4I1, COX8A, and NDUFA4). NDUFA4 encodes a subunit of the respiratory chain Complex IV. NDUFA4 dysfunction is recognized as the underlying cause of mitochondrial Complex IV deficiency nuclear type 21 (MC4DN21, OMIM 619065), a relatively mild presentation of Leigh syndrome 53 . SURF1 is another involved protein in Complex IV deficiency and is one of the most common causes of Leigh syndrome. To date, over sixty distinct SURF1 mutations have been identified as causes of SURF1-associated Leigh syndrome 54. The majority of SURF1-associated Leigh syndrome cases follow a typical course, leading to early mortality before the age of ten. However, approximately 10% of cases exhibit an atypical progression with milder symptoms and a longer life expectancy 54. Onset is usually in infancy with poor feeding and later on developmental regression. Ataxia, neuropathy ophthalmoplegia, and hypertrichosis are the most important manifestations 55. In a mice model, SURF1−/− mice exhibited lower birth weights but eventually reached body weights similar to their siblings; however, they demonstrated a mild motor delay 56. Other COX assembly factor defects associated with Leigh syndrome are deficiencies of COX10, COX15, SCO2, PET100, PET117, and TACO1 8.
Mitochondrial Complex V plays a crucial role in oxidative phosphorylation by facilitating ATP production. One of the first recognized genetic causes of Leigh syndrome was a recurrent single nucleotide mtDNA variant in the MT-ATP6 gene, with a clinical spectrum varying according to the variable mutation load 57. ATP5PO, which encodes the oligomycin sensitivity-conferring protein, is one of the proteins whose role has been recognized in Leigh syndrome presentation 58.
Other Mutations Associated with Leigh and Leigh-like Syndrome
Leigh syndrome is present in about one-third of cases of pyruvate dehydrogenase complex (PDHc) deficiency 59. Five genetic defects of pyruvate dehydrogenase (PDH) have been linked to Leigh syndrome, such as PDHA1, PDHB, PDHX, DLT, and DLD mutations. The most frequently encountered is the PDHA1 X-linked mutations 60. These disorders do not always meet the clinical and pathological features for Leigh syndrome diagnosis. Disorders of vitamins and cofactor metabolism have also been associated with Leigh syndrome, such as biotinidase deficiency, which leads to biotin-dependent enzyme impairment 61. Another treatable cause of Leigh syndrome is the SLC19A13 mutation, which leads to Wernicke-like encephalopathy presentation when not fatal 62, 63. Disorders of mitochondrial DNA maintenance associated with Leigh syndrome include biallelic pathogenetic variants of SUCLA2, SUCLG1, POLG, and RNASEH1. The most relevant ones are SUCL2 and SUCLG1 mutations, which encode two subunits of succinyl-CoA ligase (a Krebs cycle enzyme that also helps with the maintenance of mtDNA). Patients with SUCL2 variants usually have sensorineural hearing loss and dystonia, while SUCLG1 patients have systemic involvement, especially in the liver and heart 64. Many experts believe POLG mutations never meet the diagnostic criteria for Leigh syndrome, as they often present themselves with a clinically different spectrum, such as Alpers–Huttenlocher syndrome. Many other disorders of mitochondrial proteins have been linked to Leigh syndrome; among these, mutations in the SERAC1 gene lead to 3-methylglutaconic aciduria, deafness, and encephalopathy, Leigh-like (MEGDEL) syndrome, which is a disorder of lipid remodeling 65.
Coenzyme Q Deficiency
Coenzyme Q (CoQ) deficiency can have different signs and symptoms, but can resemble Leigh syndrome 66. But coenzyme Q (CoQ) deficiency should not be included in a Leigh-like syndrome per se. For example, a patient with coenzyme Q deficiency may exhibit congenital muscle weakness (hypotonia), seizures starting at age 3 months that are difficult to control and spread throughout the body, progressive muscle weakness, challenges in feeding, intermittent vomiting since age 7 months, the need for tube feeding, swelling due to low protein levels linked to poor feeding and kidney-related issues (nephrotic syndrome), ultimately resulting in death at 8 months 66. In another case involving two sisters, primary coenzyme Q deficiency presented as an adult-onset syndrome characterized by brain dysfunction, slowed growth, lack of muscle coordination (ataxia), and hearing impairment 67. Both individuals significantly improved with coenzyme Q replacement 67. Coenzyme Q10 deficiency primarily affects the brain, muscles, and kidneys due to their high energy demand. Symptoms range from severe in infancy to mild in later life. It can cause ataxia, nephrotic syndrome, and hypertrophic cardiomyopathy.
Leigh syndrome gene
Currently, over 100 nuclear DNA (nDNA) genes and defects in 16 mitochondrial DNA (mtDNA) genes encoding 10 oxidative phosphorylation (OXPHOS) subunits and 6 transfer RNAs (tRNAs) can result in Leigh syndrome 68, 31, 69, 70, 71, 5.
Commonly implicated genes in Leigh syndrome include 31, 69, 5, 72:
- Mitochondrial DNA (mtDNA) genes:
- MT-ATP6 gene which encodes for Complex V
- MT-ND genes (e.g. MT-ND5) which encode for Complex I
- Nuclear DNA (nDNA) genes:
- SURF1 gene which encodes for Complex IV
Given the wide range of genes, there is a range of inheritance patterns observed in Leigh syndrome 71, 69. This includes mitochondrial (i.e. maternal) or autosomal recessive patterns of inheritance, and very rarely, also X-linked recessive and autosomal dominant patterns of inheritance 71, 69.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Table 1. Genes involved in the Leigh Syndrome
| Inheritance | Genetic Mutations |
|---|---|
| Autosomal Recessive/ (Maternal/Sporadic) | |
| Complex I | NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NDUFA2, NDUFA9, NDUFA10, NDUFA12, NDUFA13, NDUFAF2, NDUFAF4, NDUFAF5, NDUFAF6, FOXRED1, NUBPL, NDUFAF8, TIMMDC1, NDUFB8, NDUFC2 (MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6) |
| Complex II | SDHA, SDHAF1 * |
| Complex III | UQCRQ, TTC19, BCS1L |
| Complex IV | SURF1, NDUFA4, COX4I1, COX8A, COX10, COX15, SCO2, LRPPRC, TACO1, PET100, PET117 (MT-CO1, MT-CO2, MT-CO3) |
| Complex V | ATP5MD (MT-ATP6) |
| mitochondrial DNA maintenance | POLG *, SUCLA2, SUCLG1, FBXL4, SLC25A4, SSBP1, RNASEH1, GTPBP3 |
| mitochondrial gene expression | TRMU, GTPBP3, MTFMT, EARS2, FARS2, IARS2, NARS2, PTCD3, MRPS34, GFM1, GFM2, TSFM, MTRFR, PNPT1, C12ORF65, TARS2 (MT-TI, MT-TK, MT-TL1, MT-TL2, MT-TV, MT-TW) |
| mitochondrial cofactor | PDSS2, COQ9, LIAS, LIPT1, MECR |
| mitochondrial membrane | SERAC1, MFF, SLC25A46, CLPB |
| mitochondrial toxicity | HIBCH, ECHS1, ETHE1 *, SQOR, SLC39A8, NAXE |
| mitochondrial (other) | LONP1, VPS13D, OPA1 |
| pyruvate dehydrogenase complex | PDHB, DLAT, DLD, PDHX |
| B vitamin transport and metabolism | SLC25A19, TPK1, BTD, SLC19A3 |
| miscellaneous | HPDL, ADAR, NUP62, RANBP2, MORC2 |
| Autosomal dominant | DNM1L |
| X-linked | PDHA1 *, NDUFA1, AIFM1 * |
Footnotes: * These mutations include syndromes that may resemble Leigh syndrome but show a different pathology. Ethylmalonic encephalopathy due to mutations of ETHE1 has a distinct, complex neuropathology evidenced by both autoptic and MRI examination. POLG mutations are associated with a variety of manifestations, in children in particular by Alpers–Huttenlocher disease or, later, by Spinocerebellar ataxia and epilepsy. Therefore, these mutations are not usually included in the Leigh-like syndrome spectrum. Defects in PDHA can also appear as Leigh syndrome at MRI, but they are typically associated with other lesions, such as micropore accumulation, especially in the frontal region.
[Source 73 ]Leigh syndrome inheritance pattern
In most cases, Leigh syndrome is inherited as an autosomal recessive trait. However, X-linked recessive and maternal inheritance, due to a mitochondrial DNA mutation, are additional modes of transmission.
- Leigh syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Unaffected parents are called carriers because they each carry one copy of the mutated gene and can pass it to their children.
- Leigh syndrome X-linked recessive inheritance pattern is a way a genetic trait or condition can be passed down from parent to child through mutations (changes) in a gene on the X chromosome. In males who only have one X chromosome (XY), a mutation in the copy of the gene on the single X chromosome causes the condition. Females who have two X chromosomes (XX) must have a mutation on both X chromosomes in order to be affected with the condition. If only the father or the mother has the mutated X-linked gene, the daughters are usually not affected and are called carriers because one of their X chromosomes has the mutation but the other one is normal. Sons will be affected if they inherit the mutated X-linked gene from their mother. Fathers cannot pass X-linked recessive conditions to their sons because males have XY chromosome.
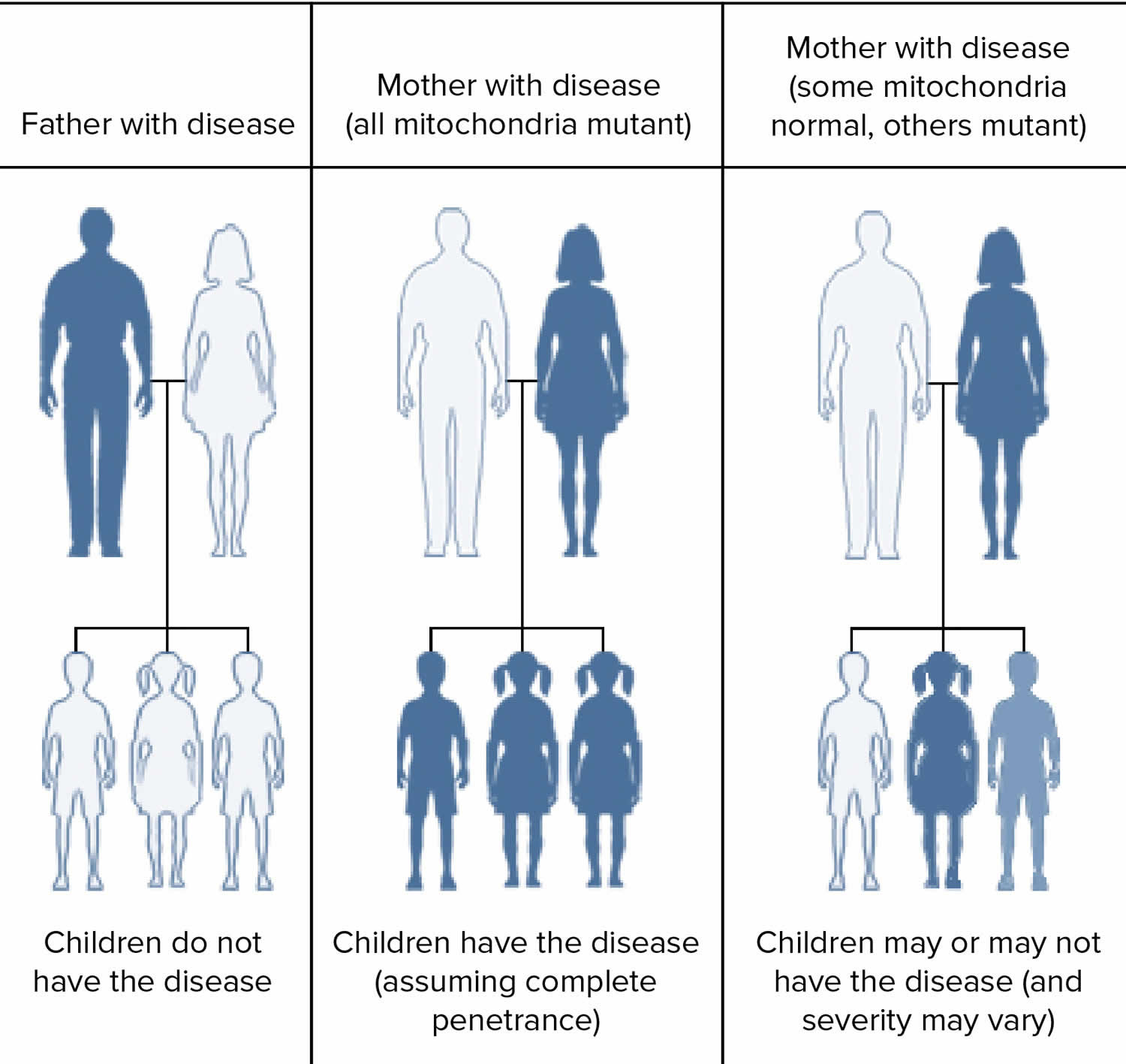
- Some Leigh syndrome is inherited in a maternal inheritance also known as mitochondrial pattern. Mitochondrial pattern of inheritance (maternal inheritance) applies to genes contained in mitochondrial DNA (mtDNA). Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 5. Leigh syndrome autosomal recessive inheritance pattern
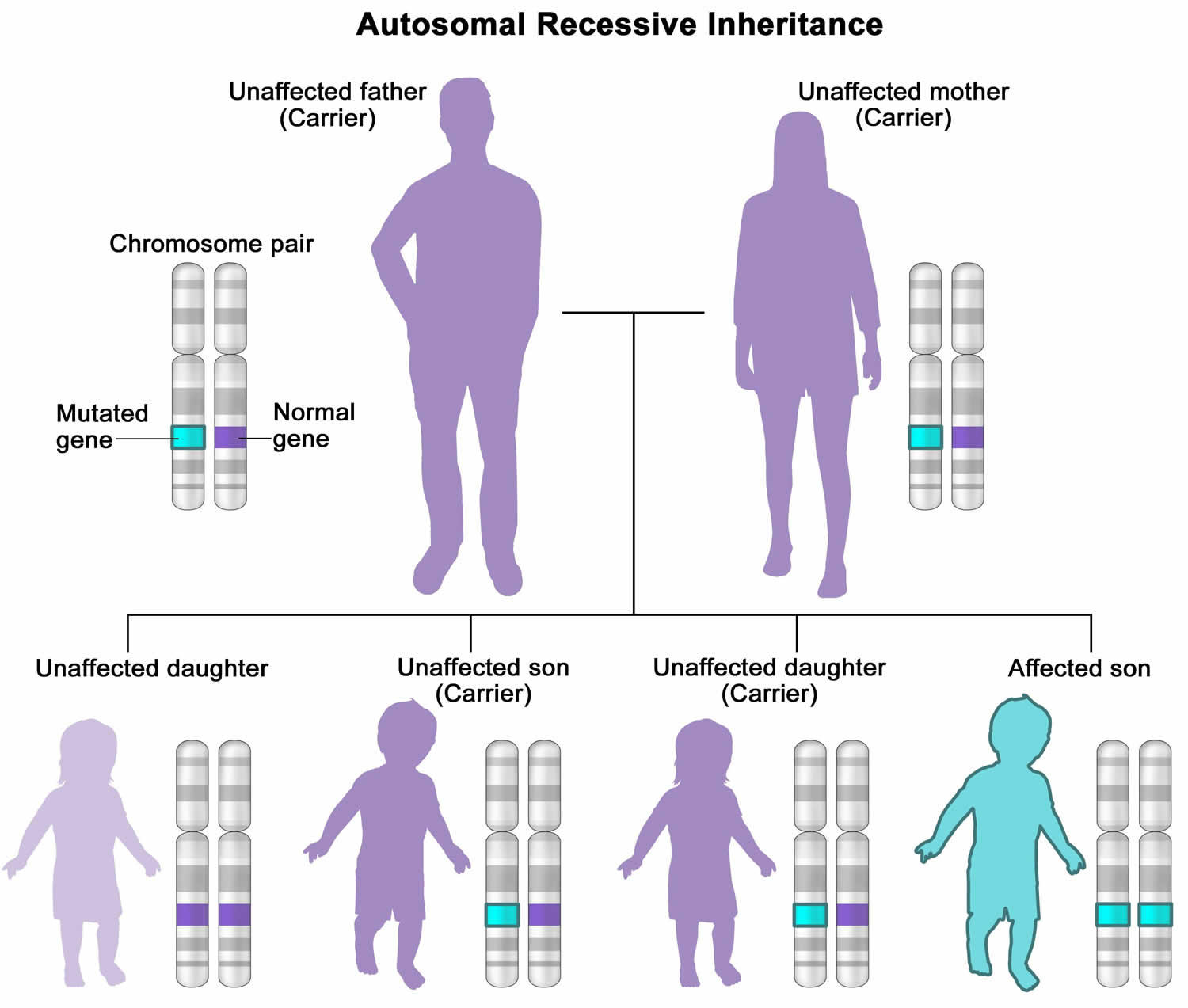
Figure 6. Leigh syndrome X-linked recessive inheritance pattern
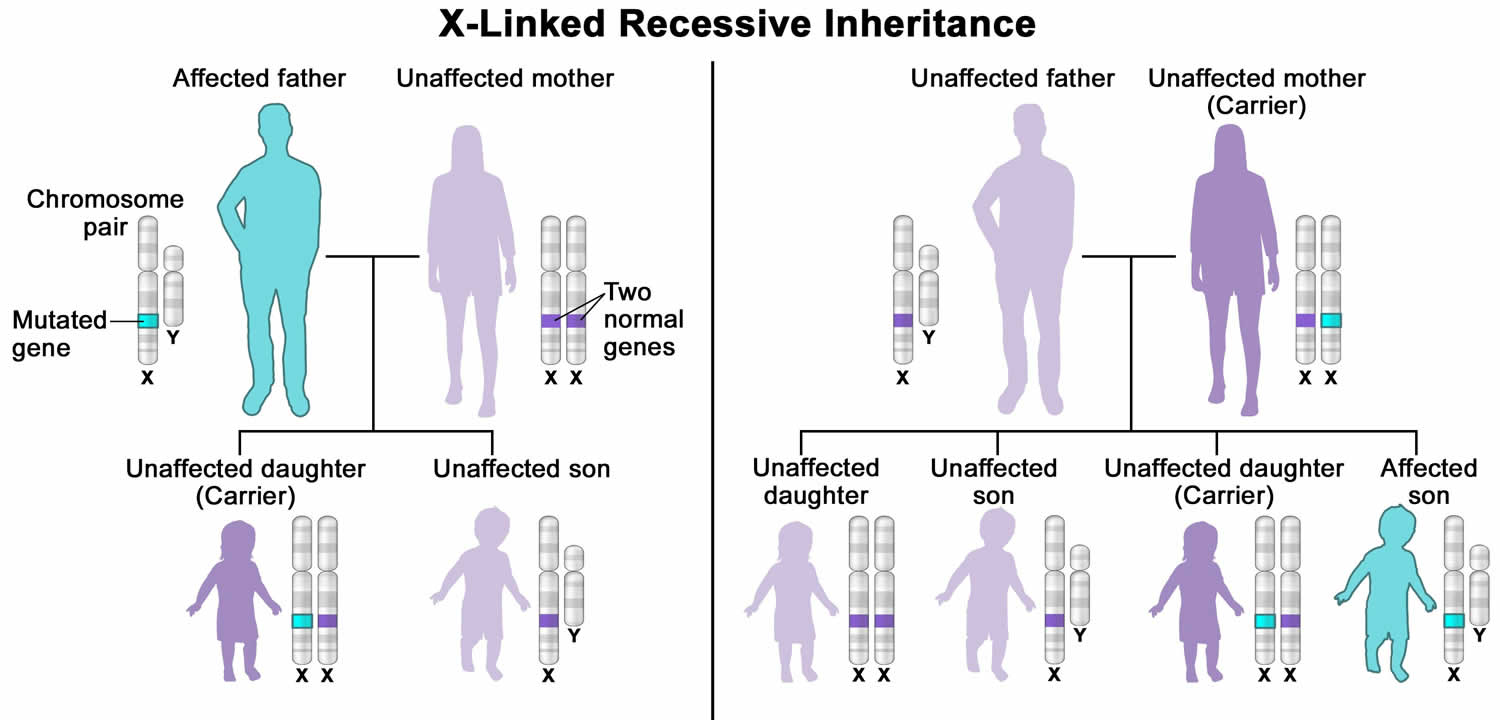
Figure 7. Leigh syndrome maternal inheritance pattern
Leigh syndrome signs and symptoms
The symptoms of classical Leigh syndrome (infantile necrotizing encephalopathy) usually begin between the ages of 3 months and 2 years. In most children, the first noticeable sign is the loss of previously acquired motor skills. When there is early onset (i.e., 3 months), loss of head control and poor sucking ability may be the first noticeable symptoms. This may be accompanied by a profound loss of appetite, recurrent vomiting, irritability, continuous crying and possible seizure activity. Delays in reaching developmental milestones may also occur. Affected infants may fail to grow and gain weight at the expected rate (failure to thrive).
If the onset of Leigh syndrome is later in childhood (e.g., 24 months), a child may experience difficulty articulating words (dysarthria) and coordinating voluntary movements such as walking or running (ataxia). Previously acquired intellectual skills may diminish and intellectual disability may also occur.
Progressive neurological deterioration associated with Leigh syndrome is marked by a variety of symptoms including generalized weakness, lack of muscle tone (hypotonia), clumsiness, tremors, muscle spasms (spasticity) that result in slow, stiff movements of the legs, and/or the absence of tendon reflexes. Further neurological development is delayed.
In addition, a substance called lactate can build up in the body, and excessive amounts are often found in the blood, urine, or the fluid that surrounds and protects the brain and spinal cord (cerebrospinal fluid [CSF]) of people with Leigh syndrome 6. Episodes of lactic acidosis may occur and are characterized by abnormally high levels of lactic acid in the blood, brain and other tissues of the body. Periodically, levels of carbon dioxide in the blood may also be abnormally elevated (hypercapnia). Lactic acidosis and hypercapnia can lead to psychomotor regression and respiratory, heart, or kidney impairment.
Children with Leigh syndrome usually develop severe breathing problems including the temporary cessation of spontaneous breathing (apnea), difficulty breathing (dyspnea), abnormally rapid breathing (hyperventilation), and/or abnormal breathing patterns (Cheyne-Stokes) and these problems can worsen until they cause acute respiratory failure 6. Some infants may also experience difficulty swallowing (dysphagia).
Many individuals with Leigh syndrome develop weakness or paralysis of the muscles that move the eyes (ophthalmoplegia); rapid, involuntary eye movements (nystagmus); strabismus (“lazy eyes” or “crossed eyes”) a condition where the eyes do not point in the same direction with one or both eyes either turning inward [esotropia], outward [exotropia], upward [hypertropia], or downward [hypotropia]; or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy) and pigmentary retinopathy leading to visual impairment leading to blindness 20, 21, 6.
Leigh syndrome may also affect the heart. Some children with this disorder may have abnormal enlargement of the heart (hypertrophic cardiomyopathy – a thickening of the heart muscle that forces the heart to work harder to pump blood), overgrowth of the fibrous membrane that divides the various chambers of the heart (asymmetric septal hypertrophy) and conduction defects such as Wolff–Parkinson–White syndrome 22, 23, 6. Disease affecting the nerves outside of the central nervous system (peripheral neuropathy) may eventually occur, causing progressive weakness of the arms and legs.
The symptoms of the X-linked infantile form of Leigh syndrome are similar to those of classical Leigh syndrome. The symptoms of the adult-onset form of Leigh syndrome (subacute necrotizing encephalomyelopathy), a very rare form of the disorder, generally begin during adolescence or early adulthood. Initial symptoms are generally related to vision and may include such abnormalities as blurred “filmy” central visual fields (central scotoma), colorblindness, and/or progressive visual loss due to degeneration of the optic nerve (bilateral optic atrophy). The neurological problems associated with the disease progress slowly in this form of the disorder. At about 50 years of age, affected individuals may find it progressively difficult to coordinate voluntary movements (ataxia). Additional late symptoms may include partial paralysis and involuntary muscle movements (spastic paresis), sudden muscle spasms (clonic jerks), grand mal seizures, and/or varying degrees of dementia.
The signs and symptoms of Leigh syndrome are caused in part by patches of damaged tissue (lesions) that develop in the brains of people with Leigh syndrome. A medical procedure called magnetic resonance imaging (MRI) reveals characteristic lesions in certain regions of the brain. These regions include the basal ganglia, which help control movement; the cerebellum, which controls the ability to balance and coordinates movement; and the brainstem, which connects the brain to the spinal cord and controls functions such as swallowing and breathing. The brain lesions are often accompanied by loss of the myelin coating around nerves (demyelination), which reduces the ability of the nerves to activate muscles used for movement or relay sensory information from the rest of the body back to the brain.
Leigh syndrome diagnosis
Consensus on Leigh syndrome diagnosis is yet to be determined; however, Leigh syndrome is suspected through the hallmarks of the disease along with findings suggestive of brainstem dysfunction in addition to T2 weighted brain MRI lesions and accessory laboratory findings 20. Brain MRI findings typically show bilateral symmetrical supra-tentorial (basal ganglia, thalamus, and sub-thalamus) and/or infra-tentorial (brainstem and dentate nuclei) lesions. A study by Ardissone et al 27 presented a predominating basal ganglia involvement of 90.2%. They also showed that both supra and infra-tentorial involvement is dominant in cases of both mtDNA (74%) and -nDNA (67%) variants, while isolated infra-tentorial variants are rare 27. Extensive research is being conducted to find genetic correlations with MRI findings of Leigh syndrome. For example, a retrospective cohort found significant associations between the SURF1 variant and inferior olivary nuclei lesions 28.
Abnormal laboratory findings may yield elevated blood, urine, and CSF lactate levels. Additional deficiencies may be observed in respiratory chain complexes through enzyme assays and pyruvate dehydrogenase complex 29. However, these laboratory findings are not consistently present. Therefore, confirmatory tests with genetic testings are required for a definitive diagnosis and the identification of specific gene mutations of Leigh syndrome 30.
Laboratory Findings
Commonly, resting levels of lactate or pyruvate in the blood are elevated 19, 74. Elevated lactate levels (hyperlactatemia) may not be initially observed at the onset of symptoms but may develop as Leigh’s disease progresses 75. Moreover, in cases where skeletal muscles are affected, levels of creatine kinase (CK) are elevated 76. Anemia has also been reported in isolated cases 75. Patients with the Faroe variant typically display elevated levels of methylmalonic acid (MMA) in their urine 7. In cases of mitochondrial Leigh syndrome, there may be increased excretion of Krebs cycle intermediates in the urine. Lactate levels may be elevated in specific patients 77. Patients with non-mitochondrial Leigh syndrome may display increased urinary excretion of 3-methylglutaconic acid or 3-methylglutaric acid [9,88]. In most cases, the levels of lactate, pyruvate, or the lactate/pyruvate ratio are elevated in the cerebrospinal fluid 19, 78, 79. In a case involving a patient with Leigh syndrome resulting from a deficiency in coenzyme Q, there was a significant reduction in coenzyme Q levels 67.
Electrophysiological Assessments
Needle electromyography can detect irregular spontaneous activity 80. Nerve-conduction studies have provided insights into potential polyneuropathy 80. Auditory-evoked brainstem potentials may exhibit extended latencies even before clinical symptoms manifest 79, 81, 82. Assessing visually evoked potentials may reveal elongation or reduction in amplitude of the P100 component 78 or in some cases, its complete absence 79. Finally, an electroencephalogram may demonstrate irregular baseline activity and focal epileptic indicators, potentially leading to secondary generalization and hypsarrhythmia 76.
Neuroimaging
When clinical evaluations and laboratory tests trigger suspicion of Leigh syndrome, a brain MRI should be obtained. Distinctive observations in patients with Leigh syndrome typically include the presence of bilateral and symmetrical hyperintensities evident in T2-weighted images. These hyperintensities are primarily located in specific brain regions, notably the basal ganglia especially the putamen and various parts of the brainstem, such as the substantia nigra, nucleus ruber, and medulla oblongata 83, 84. Other findings include microcephaly, ventricular enlargement, intracranial pseudo-cysts, and white matter abnormalities 85. Lesions in Leigh syndrome usually evolve over time; nonetheless, some case series reported partial or complete regression of lesions in follow-up MRI 86, 84. The reversibility of Leigh lesions underscores the importance of ongoing efforts to develop treatments that prevent brain damage 84. Moreover, the absence of visible lesions does not rule out a diagnosis of Leigh syndrome, as patients may develop them later in the disease course 84. It is also recommended to complement conventional MRI with proton magnetic resonance spectroscopy (MRS). The identification of a lactate peak in either brain parenchyma or cerebrospinal fluid (CSF) is considered a hallmark of mitochondrial disease.
There are correlations between MRI findings and the underlying genetic mutations in Leigh syndrome. For instance, in cases with complex I deficiency, brain MRIs often reveal bilateral symmetric brainstem lesions, at least one striatal anomaly, and an observable lactate peak in magnetic resonance spectroscopy (MRS) 79. Rarely, Leigh syndrome mimics tectal glioma, with symmetrical lesions in the midbrain and the pons, described as “giant panda” or “double panda sign” 87. Furthermore, some patients with POLG and SURF1 mutations have been reported to exhibit hypertrophic olivary degeneration, which may manifest clinically as tremors 88. POLG mutations are associated with a variety of signs and symptoms, in children in particular by Alpers–Huttenlocher disease or, later, by spinocerebellar ataxia and epilepsy. Therefore, POLG mutations are usually not included in the Leigh-like syndrome spectrum. Bindu and colleagues 89 described the presence of bilateral hypertrophic olivary degeneration on MRI in a cohort of 10 children diagnosed with Leigh and Leigh-like syndrome. Finally, the progression of Leigh syndrome over time is not always followed by a progression of lesions on MRI. Regression of lesions was demonstrated in five out of twelve patients 86. In another study, three patients had complete resolution of their lesions, demonstrating that Leigh syndrome lesions can be reversible and that the lack of lesions cannot rule out a diagnosis of Leigh syndrome 84.
Muscle Biopsies and Cultured Fibroblasts
Muscle tissue is frequently impacted in mitochondrial diseases due to its substantial energy requirements. Confirmation or exclusion of an oxidative phosphorylation (OXPHOS) disorder can be solely achieved through the analysis of a muscle biopsy 90. An essential aspect of oxidative phosphorylation (OXPHOS) deficiency diagnosis involves the biochemical assessment of muscle biopsies extracted from either the quadriceps femoris muscle or the soleus muscle. To comprehensively assess the entire oxidative phosphorylation (OXPHOS) system, immediate processing of a freshly obtained biopsy is imperative. Notably, the necessity of a muscle biopsy in the diagnostic procedure is not universal. In cases where the clinical and biochemical phenotype strongly points to a specific mutation, it is advisable to commence a genetic analysis. Another valuable diagnostic tool is the examination of cultured fibroblasts derived from skin biopsies. This approach becomes pertinent when muscle tissue accessibility is limited or when validation and clarification of muscle biopsy results are required. However, it’s worth noting that oxidative phosphorylation (OXPHOS) disorders in muscle tissue may exhibit less marked symptoms or even be absent in cultured fibroblasts.
Genetic Testing
Mitochondrial disorders occur with various clinical and biochemical features, implicating hundreds of genes present in both mtDNA and nDNA. When clinical features lack specificity or a multitude of candidate genes exist, a broader approach is necessary. A primary step in genetic diagnosis involves full sequence analysis of mtDNA, often extracted from affected tissue, primarily muscle. This approach is particularly useful for identifying mutations with high heteroplasmy levels. In cases of mtDNA depletion without detectable mutations, consideration of POLG gene mutations, encoding polymerase gamma essential for mtDNA replication and repair, becomes crucial. If mtDNA mutations are ruled out, the focus shifts to sequencing candidate nuclear genes based on the patient’s clinical and biochemical features. The conventional strategy involves sequencing the most frequently mutated genes among the selected candidates. However, nowadays next-generation sequencing (NGS) technologies enable the sequencing of multiple candidate genes or even entire exomes.
Leigh syndrome differential diagnosis
Leigh syndrome differential diagnosis may include:
- Wernicke encephalopathy also known as Wernicke syndrome and Korsakoff syndrome are related disorders that often occur due to thiamine (vitamin B1) deficiency often resulting from chronic alcohol abuse or poor nutrition. When these two disorders occur together, the term Wernicke-Korsakoff syndrome is used. In the United States, most cases occur in alcoholics. Some researchers believe Wernicke and Korsakoff syndromes are separate yet related disorders; others believe them to be different stages of the same disorder or disease spectrum. Wernicke syndrome is considered the acute phase with a shorter duration and more serious symptoms. Korsakoff syndrome is considered the chronic phase and is a long-lasting condition.
- Wernicke’s encephalopathy is a neurological disease characterized by the clinical triad of confusion, the inability to coordinate voluntary movement (ataxia), and eye (ocular) abnormalities.
- Korsakoff syndrome is a serious neurological disorder often a consequence of untreated Wernicke’s encephalopathy characterized by severe memory impairment, confusion, and confabulation (making up false memories). Korsakoff syndrome is primarily caused by thiamine (vitamin B1) deficiency, frequently due to chronic alcohol abuse.
- similar appearance but typically presents in a very different patient demographic
- mammillary bodies not typically involved in Leigh syndrome
- enhancement more common in Wernicke encephalopathy
- hemorrhagic change more common in Wernicke encephalopathy
- Other mitochondrial disorders (e.g. MEGDEL syndrome)
- brainstem and basal ganglia involvement less pronounced
- Batten disease, a rare genetic disorder, belongs to a group of progressive degenerative neurometabolic disorders known as the neuronal ceroid lipofuscinoses (NCLs). These disorders share certain similar symptoms and are distinguished in part by the age at which such symptoms appear. Batten disease is considered the juvenile form of the neuronal ceroid lipofuscinoses. The neuronal ceroid lipofuscinoses are characterized by abnormal accumulation of certain fatty, granular substances (i.e., pigmented lipids [lipopigments] ceroid and lipofuscin) within nerve cells (neurons) of the brain as well as other tissues of the body that may result in progressive deterioration (atrophy) of certain areas of the brain, neurological impairment, and other characteristic symptoms and physical findings. The symptoms of Batten disease usually become apparent between 5 and 15 years of age when progressive loss of vision, seizures, and progressive neurological degeneration develop. In some cases, initial symptoms may be more vague and include clumsiness, balance problems and behavioral or personality changes. Batten disease is inherited as an autosomal recessive trait and occurs most in families of Northern European or Scandinavian ancestry.
- Tay-Sachs disease is a rare neurodegenerative disorder caused by a deficiency in the enzyme hexosaminidase A (HEXA), leading to a buildup of fatty substances known as gangliosides in the brain and nerve cells. This abnormal accumulation of gangliosides leads to progressive dysfunction of the central nervous system. This disorder is categorized as a lysosomal storage disease. Lysosomes are the major digestive units in cells. Enzymes within lysosomes break down or “digest” nutrients, including certain complex carbohydrates and fats. Symptoms typically appear around 6 months of age and include developmental regression, vision and hearing problems, and seizures. Symptoms associated with Tay-Sachs disease may include an exaggerated startle response to sudden noises, listlessness, loss of previously acquired skills (i.e., psychomotor regression), and severely diminished muscle tone (hypotonia). With disease progression, affected infants and children may develop cherry-red spots within the middle layer of the eyes, gradual loss of vision, and deafness, increasing muscle stiffness and restricted movements (spasticity), eventual paralysis, uncontrolled electrical disturbances in the brain (seizures), and deterioration of cognitive processes (dementia). The classical form of Tay-Sachs disease occurs during infancy; an adult form (late-onset Tay-Sachs disease) may occur anytime from adolescence to the mid-30s. Tay-Sachs disease is inherited as an autosomal recessive trait. There is no cure, and the disease is usually fatal in childhood.
- NARP syndrome (Neuropathy, Ataxia, and Retinitis Pigmentosa) is a rare genetic disorder primarily affecting the nervous system. NARP syndrome (Neuropathy, Ataxia, and Retinitis Pigmentosa) is characterized by nerve disease affecting the nerves outside of the central nervous system (peripheral neuropathy), an impaired ability to coordinate voluntary movements (ataxia), an eye condition known as retinitis pigmentosa with progressive vision loss, and a variety of additional abnormalities. The specific symptoms of NARP syndrome in each individual vary greatly from case to case. Symptoms typically begin in childhood or early adulthood and can progress over time. NARP syndrome (Neuropathy, Ataxia, and Retinitis Pigmentosa) is a maternally inherited mitochondrial disease. NARP syndrome is caused by a specific mutation affecting the mitochondrial gene known as the ATPase 6 gene or MT-ATP6 gene. This mutation can also cause a specific subtype of Leigh syndrome known as maternally inherited Leigh syndrome (MILS). In fact, when individuals have more than 90 percent of mutated mitochondrial DNA (mtDNA) in their cells, they are classified as having maternally inherited Leigh syndrome (MILS) and not NARP syndrome. Most individuals with NARP syndrome have 70-80 percent of mutated mtDNA.
- Acute necrotizing encephalitis of childhood
- lactate levels are usually normal
- Biotin-thiamine-responsive basal ganglia disease
- Toxic encephalopathies (e.g. carbon monoxide poisoning)
Leigh syndrome treatment
To date, no definitive treatment has been identified for Leigh syndrome 4, 1. Current therapies include supplements with coenzyme Q10 (CoQ10) and its derivatives, thiamine (vitamin B1) (10-20 mg/kg/day), riboflavin (vitamin B2), biotin (vitamin B7) (10-15 mg/kg/day) and vitamin C, pyruvate, dichloroacetate, and a ketogenic diet 33, 34, 32, 91. In patients with SLC19A3 mutations some improvement is seen, especially in early presentation, with thiamine (10-20 mg/kg/day) and biotin (10-15 mg/kg/day) oral supplementation 91. The same is observed in high doses supplementation of thiamine (30-40 mg/kg/day) in patients with PDHA1 deficiency by stabilizing the PDHc 91, 92. Coenzyme Q10 (CoQ10) is another that is reasonably effective in mitochondrial disorders providing recovery of neurologic symptoms. Although acting in electron transfer from complexes in electron transport chain (ETC) and playing an antioxidant part in many cellular processes 93 and even higher dose are well tolerated even benefits are discussed. The recommended dose of Coenzyme Q10 (CoQ10) is 10-30 mg/kg/day via oral supplementation 91.
Table 2 below provides information on the current drugs and therapies used for Leigh syndrome and preclinical in vivo studies. However, recent systematic reviews have highlighted additional therapeutic interventions under investigation, including experimental treatments and clinical trials 94.
Table 2. Leigh syndrome treatment options
| Treatment | Specific Mutations or Deficiencies Reported |
|---|---|
| Coenzyme Q10 (CoQ10) | ND3-m.10197 G>A, Succinate: cytochrome c oxidoreductase deficiency, m.9185 T>C, m.10191 T>C, PDSS2/CoQ10 deficiency |
| EPI-743 | ND1-G3697A, SUCLA2, ETHE1 *, ND5-G13513A, EARS2, SURF1, ND1-G3697A, ND6-T14487C |
| Idebenone | Unknown |
| KH176 | Mitochondrial m.3243A>G Spectrum Disorders |
| Riboflavin | ACAD9 *- c.1240C>T |
| Thiamine | SLC19A3/Thiamine transporter-2 deficiency |
| Thiamine + Biotin + CoenzymeQ10 + Vitamin E + Vitamin C + Carnitine | SLC19A3/Thiamine transporter-2 deficiency |
| Sodium Pyruvate | Unknown, m.8993 T>G, m.9176 T>C |
| Sodium Dichloroacetate | ATP6-m.8993 T>C, ATP6-m.8993 T>G |
| N-acetylcysteine | ETHE1 * |
| Carnitine | m.8993T>C mutation |
| Ketogenic diet | PDHc deficiency, NDUFV1 |
| Plasmapheresis + IVIG | ATP6-m.9176 T>C |
| Preclinical Studies | |
| Treatment | Targeted mechanism |
| Rapamycin | Unknown, mTOR pathway |
| Hypoxia | Oxygen restriction |
| AVV gene therapy | Insertion of a target gene |
| Olaparib, veliparib | PARP inhibitors (PARPis) |
| Nicotinamide riboside and nicotinamide mononucleotide | (NAD+ precursors) enhancing SIRT1 function |
| AD4 | Lipid metabolism |
| Hispidin | MTND |
| Avanafil | MT-APT6 mutation, membrane potential |
Footnotes: * These mutations include syndromes that may resemble Leigh syndrome but show a different pathology: ACAD9 mutations are associated with a non-Leigh complex encephalopathy and often cardiomyopathy.
[Source 1 ]Coenzyme Q10 and Vatiquinone
Coenzyme Q10 (CoQ10) also called ubiquinone plays a vital role in shuttling electrons between complex II and III. In a specific case study, a patient with Leigh syndrome and an m.10197 G>A mutation experienced notable improvement after receiving coenzyme Q10 (CoQ10) supplementation for three months 95. Furthermore, an MRI scan conducted during a one-year follow-up showed the complete disappearance of the previously observed lesion. It is worth noting, however, that coenzyme Q10 (CoQ10) supplementation does not consistently produce positive results in cases where there is a coenzyme Q10 (CoQ10) deficiency 66. Six studies evaluated coenzyme Q10 (CoQ10) as a standalone treatment, with four reporting clinical improvement in patients harboring mutations in m.10197G>A, COQ2, and COQ4 96. However, it was observed that patients in advanced stages of the disease showed clinical deterioration despite coenzyme Q10 (CoQ10) initiation, underscoring the importance of early intervention 96. In some cases, kidney function improvements were noted following coenzyme Q10 (CoQ10) supplementation 97. Conversely, Scalais et al. 98 reported a case of a child who developed severe proteinuria despite receiving coenzyme Q10 (CoQ10) therapy from infancy.
Some patients with Leigh syndrome have responded positively to alternative treatments such as Vatiquinone (EPI-743) and idebenone 99. Vatiquinone (EPI-743) is a para-benzoquinone that repletes intracellular glutathione more potently than coenzyme Q10 or idebenone. Vatiquinone (EPI-743) has been tested in open-label clinical trials for mitochondrial diseases, including Leigh syndrome. Enns et al 100 found that 11 out of 12 participants showed clinical and radiological improvements. Additionally, Martinelli et al 101 observed significant improvements in 10 children with genetically confirmed Leigh syndrome treated with Vatiquinone (EPI-743). While Vatiquinone (EPI-743) appears to have broad efficacy across genetic variations, and other drugs are currently undergoing clinical trials, not all outcomes have been satisfactory 66.
Sodium Dichloroacetate and Sodium Pyruvate
Another compound, dichloroacetate (DCA), enhances pyruvate dehydrogenase (PDH) activity by reducing lactate buildup. Dichloroacetate (DCA) treatment has shown effectiveness in patients with Leigh syndrome with the T8993C mutation in ATPase 6, a subunit of complex V 102, as well as PDHc and Complex I deficiency 103. However, studies have reported mixed clinical outcomes with dichloroacetate (DCA) treatment. While it has shown potential benefits, dichloroacetate (DCA) is also associated with the risk of worsening peripheral neuropathy, even when thiamine is administered prophylactically to mitigate side effects 104. For instance, Kimura et al 102 documented successful improvement in midbrain hyperintensities in a patient with the MT-ATP6 m.8993T>C mutation following dichloroacetate (DCA) and thiamine therapy. In contrast, Koga et al 105 described a case of a child with a PDHE1 mutation who experienced clinical deterioration with dichloroacetate (DCA) but subsequently responded positively to pyruvate therapy.
Pyruvate plays a crucial role in remedying the malfunction of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) by supplying NAD, thus restoring the NADH/NAD+ ratio that becomes disrupted in disorders marked by oxidative phosphorylation due to cytochrome C oxidase deficiency 106. In a specific case study, the oral administration of sodium pyruvate over one year significantly enhanced exercise tolerance, ventricular ejection fraction, and the normalization of an abnormal echocardiographic presentation 107. Most studies focused on biochemical pyruvate dehydrogenase (PDH) deficiencies, which were not confirmed radiologically or genetically as Leigh syndrome. Only two papers met all inclusion and exclusion criteria. The highest level of evidence came from a case series by Fujii et al. 107, which included two genetically confirmed patients with Leigh syndrome with the variants m.8993T>G and m.9176T>C. Both patients, who were bedbound, showed improvements in the Newcastle Pediatric Mitochondrial Disease Scale (NPMDS) following pyruvate treatment 108.
Sonlicromanol
Sonlicromanol (KH176) is a promising redox-modulating agent under investigation for mitochondrial diseases, including Leigh syndrome. A Phase 1 study 109 evaluating Sonlicromanol (KH176) in patients with various mitochondrial disorders, including Leigh syndrome, demonstrated that the agent was generally well tolerated. However, at higher doses, it was associated with QTc prolongation and T-wave morphological changes, necessitating careful dose optimization and monitoring 110. Building on these findings, randomized, multi-center Phase 2 trials are currently recruiting participants with a range of mitochondrial disorders, such as MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), MIDD (maternally inherited diabetes and deafness), Leigh syndrome, mitochondrial myopathies, and mitochondrial encephalopathies. These studies aim to evaluate further the safety and efficacy of Sonlicromanol (KH176) across different mitochondrial disease phenotypes. Notably, a safety and efficacy trial focusing on m.3243A>G-associated mitochondrial disease has already been completed, providing valuable insights into the therapeutic potential of Sonlicromanol (KH176) in this specific genetic context 111.
Vitamins Supplementation
Vitamin supplementation is another approach to minimixe the neurological symptoms of Leigh syndrome. High doses of Riboflavin (vitamin B2) enhance muscle strength and alleviate lactic acidosis in the context of complex I deficiency due to mutation in ACAD9 mutation, which is a crucial factor for the operation of the oxidative phosphorylation (OXPHOS) system and ATP production 111, 112. ACAD9 mutation seems to show a different pathology from the one observed in Leigh syndrome; ACAD9 mutations are associated with a non-Leigh complex encephalopathy and often cardiomyopathy in most cases. Riboflavin (vitamin B2) was utilized in 18 studies, though it was predominantly administered as part of a “mitochondrial cocktail” rather than as a standalone treatment. Only three studies specifically evaluated riboflavin monotherapy. Two studies demonstrated clinical improvement in patients with ACAD9 mutations 111, 113. Notably, Gerards et al 112 reported the restoration of complex I activity in a family with a homozygous ACAD9 (c.1594C>T) mutation following riboflavin (vitamin B2) monotherapy.
Moreover, early supplementation with both thiamine (vitamin B1) and biotin (vitamin B7) in patients with Leigh syndrome caused by a mutation in SLC19A3, a gene essential to thiamine transporter-2 deficiency, has been found to have an immediate therapeutic effect 114, 115. The SLC19A3 gene encodes a thiamine transporter, and mutations in SLC19A3 gene are associated with Biotin-Responsive Basal Ganglia Disease (BBGD). A review of 22 and 57 studies revealed that biotin and thiamine supplementation are commonly used as part of a “mitochondrial cocktail” for treating mitochondrial disorders. However, only two case series have directly compared biotin or thiamine monotherapy versus combination therapy. Debs et al 115 reported clinical and radiological improvement in two siblings with SLC19A3 missense mutations, both with biotin (vitamin B7) monotherapy and combination therapy. In contrast, Tabarki et al 116 found no significant difference in the Burke–Fahn–Marsden Dystonia Rating Scale (BFMDRS) scores between thiamine (vitamin B1) monotherapy and combination therapy in an open-label prospective study involving 20 children with the SLC19A3 c.1264A>G (p.Thr422Ala) mutation. However, combination therapy with both thiamine (vitamin B1) and biotin (vitamin B7) was associated with a significantly faster recovery from acute crises (2 days vs. 3 days) compared to monotherapy 117.
N-Acetylcysteine and Carnitine
N-acetylcysteine (NAC) is a compound that serves as a precursor to glutathione, a powerful antioxidant that helps neutralize toxic sulfides 118. In one study, Viscomi et al 118 reported positive outcomes in five children with homozygous 505 + 1G>T splice-site mutations in the ETHE1 gene, responsible for a syndrome that shows a different pathology but shares many similarities with Leigh syndrome. These children experienced a reduced seizure frequency, neurological improvements, and decreased symptoms such as acrocyanosis (bluish discoloration of extremities) and petechiae (small red or purple spots on the skin). Additionally, their episodes of diarrhea improved, which was likely enhanced by the combined use of Metronidazole, a bactericidal and prokinetic agent 119. Another case, described by Shayota et al 119, documented developmental improvements in a patient with an ECHS1 mutation following N-acetylcysteine (NAC) treatment. However, the patient was also on a valine-restricted diet, making it difficult to determine the exact contribution of N-acetylcysteine (NAC) to the observed progress 120. While these findings are encouraging, further studies are needed to clarify N-acetylcysteine (NAC)’s role in managing mitochondrial disorders and to determine its effectiveness as a standalone treatment.
Carnitine plays a crucial role in fatty acid oxidation. It is commonly included in mitochondrial cocktail therapies with combinations of nutraceuticals, cofactors, and antioxidants to support mitochondrial function and bypass defects in the electron transport chain 121. However, Carnitine effectiveness remains uncertain, as many case series and reports have documented its use with little to no noticeable benefit 96. One notable case, described by Toth et al 121 involved a patient with the m.8993T>C mutation who experienced sudden loss of mobility following an upper respiratory tract infection. The patient’s condition improved after receiving carnitine supplementation, but it is unclear whether this was due to the treatment itself or simply the natural resolution of the infection. The authors noted that the patient had low plasma and muscle carnitine levels before treatment. Later, at age 13, the patient experienced worsening ataxia and muscle weakness, which improved after the carnitine dose was adjusted based on body weight 122. While carnitine remains a widely used supplement in mitochondrial disease management, further studies are needed to determine its actual therapeutic value, particularly regarding disease progression and symptomatic relief.
Ketogenic Diet
Ketogenic Diet seems to extend longevity and ameliorate mental problems 123. Moreover, it appears to reverse the oculomotor palsy of Leigh syndrome caused by the pathogenic NDUFV1 variant. Ketogenic diets are believed to enhance fatty acid beta-oxidation, providing an alternative energy pathway when oxidative phosphorylation is impaired 124. These diets have shown effectiveness in controlling epilepsy, improving eye movements, and supporting mental development in patients with mutations in genes such as ECHS1, POLG, TMEM126B, and PDHA 1, which causes pyruvate dehydrogenase deficiency 96. Additionally, protein and valine-restricted diets have been reported to benefit patients with mutations affecting valine degradation pathways, such as those in HIBCH and ECHS1 124. These dietary interventions prevent the buildup of toxic metabolites like methacrylyl-CoA and acryloyl-CoA 120. However, ketogenic diets can also pose risks, such as inducing metabolic acidosis and potentially worsening clinical symptoms in some patients with Leigh syndrome 1.
Leigh syndrome prognosis
Despite progress in medical treatments, the prognosis (outlook) for individuals with Leigh syndrome remains poor with death usually occurring in childhood often occurring before the age of five. The later the onset of Leigh syndrome, the slower the deterioration. Death is most frequently due to respiratory failure 125.
Factors associated with a worse prognosis of Leigh syndrome are 31, 126:
- Leigh syndrome onset before 6 months of age
- Admission to an intensive care unit (ICU) due to acute exacerbations
- Brainstem lesions
- Magnetic resonance spectroscopy (MRS) of the brain reveal lactate peak
- Delayed diagnosis also contributes to poor prognosis 127.
Nevertheless, by implementing monitoring of patients who exhibit respiratory problems and employing a range of diagnostic assessments to assess brainstem function, including magnetic resonance imaging (MRI), auditory-evoked brainstem potentials, somatosensory-evoked potentials, blink reflex, or polysomnography, there is the potential to prevent sudden and early fatalities in individuals with early-onset Leigh syndrome 1. However, most of the treatments are palliative and not radical treatments 1. Among current treatments, adeno-associated virus (AAV) recombinant vectors hold promise for the development of targeted therapies and, as of now, represent the only possibility for a cure as they convey the missing gene to the brain 128, 129. However, more clinical studies are needed to evaluate the safety and efficacy of gene therapy.
- Magro G, Laterza V, Tosto F. Leigh Syndrome: A Comprehensive Review of the Disease and Present and Future Treatments. Biomedicines. 2025 Mar 17;13(3):733. doi: 10.3390/biomedicines13030733[↩][↩][↩][↩][↩][↩][↩]
- Schubert Baldo M, Vilarinho L. Molecular basis of Leigh syndrome: a current look. Orphanet J Rare Dis. 2020 Jan 29;15(1):31. doi: 10.1186/s13023-020-1297-9. Erratum in: Orphanet J Rare Dis. 2020 Mar 25;15(1):77. doi: 10.1186/s13023-020-1351-7[↩][↩]
- Saini A.G., Chatterjee D., Bhagwat C., Vyas S., Attri S.V. Leigh syndrome in an infant: Autopsy and histopathology findings. Autops. Case Rep. 2021;11:e2021334. doi: 10.4322/acr.2021.334[↩][↩]
- Chen L., Cui Y., Jiang D., Ma C.Y., Tse H.-F., Hwu W.-L., Lian Q. Management of Leigh syndrome: Current status and new insights. Clin. Genet. 2018;93:1131–1140. doi: 10.1111/cge.13139[↩][↩]
- Ardissone A, Bruno C, Diodato D, et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: a study from the Italian network of mitochondrial diseases. Orphanet J Rare Dis. 2021 Oct 9;16(1):413. doi: 10.1186/s13023-021-02029-3[↩][↩][↩]
- Leigh syndrome. https://medlineplus.gov/genetics/condition/leigh-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Rahman S., Blok R.B., Dahl H.H., Danks D.M., Kirby D.M., Chow C.W., Christodoulou J., Thorburn D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996;39:343–351. doi: 10.1002/ana.410390311[↩][↩]
- Rahman S. Leigh syndrome. Handb Clin Neurol. 2023;194:43-63. doi: 10.1016/B978-0-12-821751-1.00015-4[↩][↩][↩][↩][↩][↩][↩]
- Leigh D. Subacute necrotizing Encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14(3):216–221. doi: 10.1136/jnnp.14.3.216[↩][↩]
- Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJ, Mayatepek E, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):257–265. doi: 10.1136/jnnp-2012-304426[↩][↩]
- Schubert Baldo M., Vilarinho L. Molecular basis of Leigh syndrome: A current look. Orphanet J. Rare Dis. 2020;15:31. doi: 10.1186/s13023-020-1297-9[↩]
- Fassone E, Rahman S. Complex I deficiency: clinical features, biochemistry and molecular genetics. J Med Genet. 2012;49(9):578–590. doi: 10.1136/jmedgenet-2012-101159[↩]
- LEIGH SYNDROME, NUCLEAR; NULS. https://omim.org/entry/256000[↩]
- LEIGH SYNDROME, MITOCHONDRIAL; MILS. https://omim.org/entry/500017[↩]
- Baertling F., Rodenburg R.J., Schaper J., Smeitink J.A., Koopman W.J., Mayatepek E., Morava E., Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426[↩]
- Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr. Neurol. 2008;39:223–235. doi: 10.1016/j.pediatrneurol.2008.07.013[↩]
- Di Rocco M., Caruso U., Moroni I., Lupino S., Lamantea E., Fantasia A.R., Borrone C., Gibson K.M. 3-Methylglutaconic aciduria and hypermethioninaemia in a child with clinical and neuroradiological findings of Leigh disease. J. Inherit. Metab. Dis. 1999;22:593–598. doi: 10.1023/A:1005565610613[↩]
- Cooper M.P., Qu L., Rohas L.M., Lin J., Yang W., Erdjument-Bromage H., Tempst P., Spiegelman B.M. Defects in energy homeostasis in Leigh syndrome French Canadian variant through PGC-1alpha/LRP130 complex. Genes Dev. 2006;20:2996–3009. doi: 10.1101/gad.1483906[↩]
- Chol M., Lebon S., Bénit P., Chretien D., de Lonlay P., Goldenberg A., Odent S., Hertz-Pannier L., Vincent-Delorme C., Cormier-Daire V., et al. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J. Med. Genet. 2003;40:188–191. doi: 10.1136/jmg.40.3.188[↩][↩][↩]
- Chang X., Wu Y., Zhou J., Meng H., Zhang W., Guo J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine. 2020;99:e18634. doi: 10.1097/MD.0000000000018634[↩][↩][↩][↩][↩]
- Han J., Lee Y.-M., Kim S.M., Han S.Y., Lee J.B. Ophthalmological manifestations in patients with Leigh syndrome. Br. J. Ophthalmol. 2015;99:528–535. doi: 10.1136/bjophthalmol-2014-305704[↩][↩]
- Sofou K., de Coo I.F.M., Ostergaard E., Isohanni P., Naess K., De Meirleir L., Tzoulis C., Uusimaa J., Lönnqvist T., Bindoff L.A., et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018;55:21–27. doi: 10.1136/jmedgenet-2017-104891[↩][↩][↩][↩]
- Brecht M., Richardson M., Taranath A., Grist S., Thorburn D., Bratkovic D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015;19:95–100. doi: 10.1007/8904_2014_375[↩][↩]
- Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. doi: 10.1002/ana.24551[↩][↩]
- Kistol D., Tsygankova P., Krylova T., Bychkov I., Itkis Y., Nikolaeva E., Mikhailova S., Sumina M., Pechatnikova N., Kurbatov S., et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023;24:1597. doi: 10.3390/ijms24021597[↩][↩]
- Alves C.A.P.F., Teixeira S.R., Martin-Saavedra J.S., Guimarães Gonçalves F., Lo Russo F., Muraresku C., McCormick E.M., Falk M.J., Zolkipli-Cunningham Z., Ganetzky R., et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020;88:218–232. doi: 10.1002/ana.25789[↩][↩]
- Ardissone A., Bruno C., Diodato D., Donati A., Ghezzi D., Lamantea E., Lamperti C., Mancuso M., Martinelli D., Primiano G., et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: A study from the Italian network of mitochondrial diseases. Orphanet J. Rare Dis. 2021;16:413. doi: 10.1186/s13023-021-02029-3[↩][↩][↩][↩]
- Alves C.A.P.F., Teixeira S.R., Martin-Saavedra J.S., Gonçalves F.G., Russo F.L., Muraresku C., McCormick E.M., Falk M.J., Zolkipli-Cunningham Z., Ganetzky R., et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020;88:218–232. doi: 10.1002/ana.25789[↩][↩]
- Baertling F., Rodenburg R.J., Schaper J., Smeitink J.A., Koopman W.J.H., Mayatepek E., Morava E., Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426[↩][↩]
- Ogawa E., Shimura M., Fushimi T., Tajika M., Ichimoto K., Matsunaga A., Tsuruoka T., Ishige M., Fuchigami T., Yamazaki T., et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017;40:685–693. doi: 10.1007/s10545-017-0042-6[↩][↩]
- Chang X, Wu Y, Zhou J, Meng H, Zhang W, Guo J. A Meta-Analysis and Systematic Review of Leigh Syndrome: Clinical Manifestations, Respiratory Chain Enzyme Complex Deficiency, and Gene Mutations. Medicine. 2020;99(5):e18634. doi:10.1097/md.0000000000018634[↩][↩][↩][↩]
- Baertling F, Rodenburg RJ, Schaper J, Smeitink JA, Koopman WJ, Mayatepek E, Morava E, Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry. 2014 Mar;85(3):257-65. doi: 10.1136/jnnp-2012-304426[↩][↩][↩]
- Goldenberg PC, Steiner RD, Merkens LS, Dunaway T, Egan RA, Zimmerman EA, Nesbit G, Robinson B, Kennaway NG. Remarkable improvement in adult Leigh syndrome with partial cytochrome c oxidase deficiency. Neurology. 2003 Mar 11;60(5):865-8. doi: 10.1212/01.wnl.0000049460.72439.7f[↩][↩]
- Avula S, Parikh S, Demarest S, Kurz J, Gropman A. Treatment of mitochondrial disorders. Curr Treat Options Neurol. 2014 Jun;16(6):292. doi: 10.1007/s11940-014-0292-7[↩][↩]
- Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011;108:13534–9. doi: 10.1073/pnas.1109263108[↩]
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.[↩]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.[↩]
- Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res. 2015 Jan;141(1):13-26. doi: 10.4103/0971-5916.154489[↩][↩]
- Van de Wal M.A.E., Adjobo-Hermans M.J.W., Keijer J., Schirris T.J.J., Homberg J.R., Wieckowski M.R., Grefte S., van Schothorst E.M., van Karnebeek C., Quintana A., et al. Ndufs4 knockout mouse models of Leigh syndrome: Pathophysiology and intervention. Brain. 2022;145:45–63. doi: 10.1093/brain/awab426[↩][↩]
- Calvaruso M.A., Willems P., van den Brand M., Valsecchi F., Kruse S., Palmiter R., Smeitink J., Nijtmans L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 2012;21:115–120. doi: 10.1093/hmg/ddr446[↩]
- Liu L., Zhang K., Sandoval H., Yamamoto S., Jaiswal M., Sanz E., Li Z., Hui J., Graham B.H., Quintana A., et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019[↩]
- Procaccio V., Wallace D.C. Late-onset Leigh syndrome in a patient with mitochondrial complex I NDUFS8 mutations. Neurology. 2004;62:1899–1901. doi: 10.1212/01.WNL.0000125251.56131.65[↩][↩]
- Wang S., Kang Y., Wang R., Deng J., Yu Y., Yu J., Wang J. Emerging Roles of NDUFS8 Located in Mitochondrial Complex I in Different Diseases. Molecules. 2022;27:8754. doi: 10.3390/molecules27248754[↩]
- Kirby D.M., Kahler S.G., Freckmann M.L., Reddihough D., Thorburn D.R. Leigh disease caused by the mitochondrial DNA G14459A mutation in unrelated families. Ann. Neurol. 2000;48:102–104. doi: 10.1002/1531-8249(200007)48:1<102::AID-ANA15>3.0.CO;2-M[↩]
- Nesbitt V., Morrison P.J., Crushell E., Donnelly D.E., Alston C.L., He L., McFarland R., Taylor R.W. The clinical spectrum of the m.10191T>C mutation in complex I-deficient Leigh syndrome. Dev. Med. Child Neurol. 2012;54:500–506. doi: 10.1111/j.1469-8749.2012.04224.x[↩]
- Rustin P., Rötig A. Inborn errors of complex II—Unusual human mitochondrial diseases. Biochim. Biophys. Acta. 2002;1553:117–122. doi: 10.1016/S0005-2728(01)00228-6[↩]
- Alston C.L., Davison J.E., Meloni F., van der Westhuizen F.H., He L., Hornig-Do H.T., Peet A.C., Gissen P., Goffrini P., Ferrero I., et al. Recessive germline SDHA and SDHB mutations causing leukodystrophy and isolated mitochondrial complex II deficiency. J. Med. Genet. 2012;49:569–577. doi: 10.1136/jmedgenet-2012-101146[↩]
- Brockmann K., Bjornstad A., Dechent P., Korenke C.G., Smeitink J., Trijbels J.M.F., Athanassopoulos S., Villagran R., Skjeldal O.H., Wilichowski E., et al. Succinate in dystrophic white matter: A proton magnetic resonance spectroscopy finding characteristic for complex II deficiency. Ann. Neurol. 2002;52:38–46. doi: 10.1002/ana.10232[↩]
- Atwal P.S. Mutations in the Complex III Assembly Factor Tetratricopeptide 19 Gene TTC19 Are a Rare Cause of Leigh Syndrome. JIMD Rep. 2014;14:43–45. doi: 10.1007/8904_2013_282[↩]
- Koch J., Feichtinger R.G., Freisinger P., Pies M., Schrödl F., Iuso A., Sperl W., Mayr J.A., Prokisch H., Haack T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016;53:270–278. doi: 10.1136/jmedgenet-2015-103500[↩]
- Spinazzi M., Radaelli E., Horré K., Arranz A.M., Gounko N.V., Agostinis P., Maia T.M., Impens F., Morais V.A., Lopez-Lluch G., et al. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA. 2019;116:277–286. doi: 10.1073/pnas.1811938116[↩]
- Barel O., Shorer Z., Flusser H., Ofir R., Narkis G., Finer G., Shalev H., Nasasra A., Saada A., Birk O.S. Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ. Am. J. Hum. Genet. 2008;82:1211–1216. doi: 10.1016/j.ajhg.2008.03.020[↩]
- Misceo D., Strømme P., Bitarafan F., Chawla M.S., Sheng Y., Bach de Courtade S.M., Eide L., Frengen E. Biallelic NDUFA4 Deletion Causes Mitochondrial Complex IV Deficiency in a Patient with Leigh Syndrome. Genes. 2024;15:500. doi: 10.3390/genes15040500[↩]
- Lee I.C., Chiang K.L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants. 2021;10:1950. doi: 10.3390/antiox10121950[↩][↩]
- Wedatilake Y., Brown R.M., McFarland R., Yaplito-Lee J., Morris A.A., Champion M., Jardine P.E., Clarke A., Thorburn D.R., Taylor R.W., et al. SURF1 deficiency: A multi-centre natural history study. Orphanet J. Rare Dis. 2013;8:96. doi: 10.1186/1750-1172-8-96[↩]
- Bartke A. New findings in gene knockout, mutant and transgenic mice. Exp. Gerontol. 2008;43:11–14. doi: 10.1016/j.exger.2007.10.009[↩]
- Ball M, Thorburn DR, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome Spectrum. 2003 Oct 30 [Updated 2024 May 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1173[↩]
- Ganapathi M., Friocourt G., Gueguen N., Friederich M.W., Le Gac G., Okur V., Loaëc N., Ludwig T., Ka C., Tanji K., et al. A homozygous splice variant in ATP5PO, disrupts mitochondrial complex V function and causes Leigh syndrome in two unrelated families. J. Inherit. Metab. Dis. 2022;45:996–1012. doi: 10.1002/jimd.12526[↩]
- Patel K.P., O’Brien T.W., Subramony S.H., Shuster J., Stacpoole P.W. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Mol. Genet. Metab. 2012;106:385–394. doi: 10.1016/j.ymgme.2012.03.017[↩]
- Quinonez SC, Thoene JG. Dihydrolipoamide Dehydrogenase Deficiency. 2014 Jul 17 [Updated 2021 Sep 30]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK220444[↩]
- Mitchell G., Ogier H., Munnich A., Saudubray J.M., Shirrer J., Charpentier C., Rocchiccioli F. Neurological Deterioration and Lactic Acidemia in Biotinidase Deficiency. A Treatable Cond. Mimicking Leigh’s Dis. 1986;17:129–131. doi: 10.1055/s-2008-1052513[↩]
- Gerards M., Kamps R., van Oevelen J., Boesten I., Jongen E., de Koning B., Scholte H.R., de Angst I., Schoonderwoerd K., Sefiani A., et al. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain. 2013;136:882–890. doi: 10.1093/brain/awt013[↩]
- Fassone E., Rahman S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012;49:578–590. doi: 10.1136/jmedgenet-2012-101159[↩]
- Carrozzo R., Verrigni D., Rasmussen M., de Coo R., Amartino H., Bianchi M., Buhas D., Mesli S., Naess K., Born A.P., et al. Succinate-CoA ligase deficiency due to mutations in SUCLA2 and SUCLG1: Phenotype and genotype correlations in 71 patients. J. Inherit. Metab. Dis. 2016;39:243–252. doi: 10.1007/s10545-015-9894-9[↩]
- Wortmann S.B., van Hasselt P.M., Barić I., Burlina A., Darin N., Hörster F., Coker M., Ucar S.K., Krumina Z., Naess K., et al. Eyes on MEGDEL: Distinctive basal ganglia involvement in dystonia deafness syndrome. Neuropediatrics. 2015;46:98–103. doi: 10.1055/s-0034-1399755[↩]
- López L.C., Schuelke M., Quinzii C.M., Kanki T., Rodenburg R.J., Naini A., Dimauro S., Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am. J. Hum. Genet. 2006;79:1125–1129. doi: 10.1086/510023[↩][↩][↩][↩]
- Van Maldergem L., Trijbels F., DiMauro S., Sindelar P.J., Musumeci O., Janssen A., Delberghe X., Martin J.J., Gillerot Y. Coenzyme Q-responsive Leigh’s encephalopathy in two sisters. Ann. Neurol. 2002;52:750–754. doi: 10.1002/ana.10371[↩][↩][↩]
- Rahman J, Noronha A, Thiele I, Rahman S. Leigh map: a novel computational diagnostic resource for mitochondrial disease. Ann Neurol. 2017;81(1):9–16. doi: 10.1002/ana.24835[↩]
- Bakare A, Lesnefsky E, Iyer S. Leigh Syndrome: A Tale of Two Genomes. Front Physiol. 2021;12:693734. doi:10.3389/fphys.2021.693734[↩][↩][↩][↩]
- Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion. 2008 Dec;8(5-6):396-413. doi: 10.1016/j.mito.2008.05.003[↩]
- Ruhoy IS, Saneto RP. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl Clin Genet. 2014 Nov 13;7:221-34. doi: 10.2147/TACG.S46176[↩][↩][↩]
- Medina L, Chi TL, DeVivo DC, Hilal SK. MR findings in patients with subacute necrotizing encephalomyelopathy (Leigh syndrome): correlation with biochemical defect. AJR Am J Roentgenol. 1990 Jun;154(6):1269-74. doi: 10.2214/ajr.154.6.2159689[↩]
- Rahman S, Thorburn DR, Ball M. Nuclear Gene-Encoded Leigh Syndrome Spectrum Overview. 2015 Oct 1 [Updated 2025 May 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK320989[↩]
- Yang Y.L., Sun F., Zhang Y., Qian N., Yuan Y., Wang Z.X., Qi Y., Xiao J.X., Wang X.Y., Qi Z.Y., et al. Clinical and laboratory survey of 65 Chinese patients with Leigh syndrome. Chin. Med. J. 2006;119:373–377. doi: 10.1097/00029330-200603010-00004[↩]
- Bénit P., Chretien D., Kadhom N., de Lonlay-Debeney P., Cormier-Daire V., Cabral A., Peudenier S., Rustin P., Munnich A., Rötig A. Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet. 2001;68:1344–1352. doi: 10.1086/320603[↩][↩]
- Horváth R., Abicht A., Holinski-Feder E., Laner A., Gempel K., Prokisch H., Lochmüller H., Klopstock T., Jaksch M. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA) J. Neurol. Neurosurg. Psychiatry. 2006;77:74–76. doi: 10.1136/jnnp.2005.067041[↩][↩]
- Lebon S., Chol M., Benit P., Mugnier C., Chretien D., Giurgea I., Kern I., Girardin E., Hertz-Pannier L., de Lonlay P., et al. Recurrent de novo mitochondrial DNA mutations in respiratory chain deficiency. J. Med. Genet. 2003;40:896–899. doi: 10.1136/jmg.40.12.896[↩]
- Martín M.A., Blázquez A., Gutierrez-Solana L.G., Fernández-Moreira D., Briones P., Andreu A.L., Garesse R., Campos Y., Arenas J. Leigh syndrome associated with mitochondrial complex I deficiency due to a novel mutation in the NDUFS1 gene. Arch. Neurol. 2005;62:659–661. doi: 10.1001/archneur.62.4.659[↩][↩]
- Malfatti E., Bugiani M., Invernizzi F., de Souza C.F., Farina L., Carrara F., Lamantea E., Antozzi C., Confalonieri P., Sanseverino M.T., et al. Novel mutations of ND genes in complex I deficiency associated with mitochondrial encephalopathy. Brain. 2007;130:1894–1904. doi: 10.1093/brain/awm114[↩][↩][↩][↩]
- Rossi A, Biancheri R, Bruno C, Di Rocco M, Calvi A, Pessagno A, Tortori-Donati P. Leigh Syndrome with COX deficiency and SURF1 gene mutations: MR imaging findings. AJNR Am J Neuroradiol. 2003 Jun-Jul;24(6):1188-91. https://pmc.ncbi.nlm.nih.gov/articles/PMC8148997[↩][↩]
- Loeffen J., Smeitink J., Triepels R., Smeets R., Schuelke M., Sengers R., Trijbels F., Hamel B., Mullaart R., van den Heuvel L. The first nuclear-encoded complex I mutation in a patient with Leigh syndrome. Am. J. Hum. Genet. 1998;63:1598–1608. doi: 10.1086/302154[↩]
- Araki S., Hayashi M., Yasaka A., Maruki K. Electrophysiological brainstem dysfunction in a child with Leigh disease. Pediatr. Neurol. 1997;16:329–333. doi: 10.1016/S0887-8994(97)00020-9[↩]
- Martin E., Burger R., Wiestler O.D., Caduff R., Boltshauser E., Boesch C. Brainstem lesion revealed by MRI in a case of Leigh’s disease with respiratory failure. Pediatr. Radiol. 1990;20:349–350. doi: 10.1007/BF02013174[↩]
- Bonfante E., Koenig M.K., Adejumo R.B., Perinjelil V., Riascos R.F. The neuroimaging of Leigh syndrome: Case series and review of the literature. Pediatr. Radiol. 2016;46:443–451. doi: 10.1007/s00247-015-3523-5[↩][↩][↩][↩][↩]
- Gerards M., Sallevelt S.C.E.H., Smeets H.J.M. Leigh syndrome: Resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol. Genet. Metab. 2016;117:300–312. doi: 10.1016/j.ymgme.2015.12.004[↩]
- Sofou K., Steneryd K., Wiklund L.M., Tulinius M., Darin N. MRI of the brain in childhood-onset mitochondrial disorders with central nervous system involvement. Mitochondrion. 2013;13:364–371. doi: 10.1016/j.mito.2013.04.008[↩][↩]
- Rio M., Lebre A.S., de Lonlay P., Valayannopoulos V., Desguerre I., Dufier J.L., Grévent D., Zilbovicius M., Tréguier C., Brunelle F., et al. Mitochondrial ND5 mutations mimicking brainstem tectal glioma. Neurology. 2010;75:93. doi: 10.1212/WNL.0b013e3181e6214a[↩]
- Merante F, Petrova-Benedict R, MacKay N, Mitchell G, Lambert M, Morin C, De Braekeleer M, Laframboise R, Gagné R, Robinson BH. A biochemically distinct form of cytochrome oxidase (COX) deficiency in the Saguenay-Lac-Saint-Jean region of Quebec. Am J Hum Genet. 1993 Aug;53(2):481-7. https://pmc.ncbi.nlm.nih.gov/articles/instance/1682348/pdf/ajhg00053-0187.pdf[↩]
- Bindu P.S., Taly A.B., Sonam K., Govindaraju C., Arvinda H.R., Gayathri N., Bharath M.M., Ranjith D., Nagappa M., Sinha S., et al. Bilateral hypertrophic olivary nucleus degeneration on magnetic resonance imaging in children with Leigh and Leigh-like syndrome. Br. J. Radiol. 2014;87:20130478. doi: 10.1259/bjr.20130478[↩]
- Morin C., Mitchell G., Larochelle J., Lambert M., Ogier H., Robinson B.H., De Braekeleer M. Clinical, metabolic, and genetic aspects of cytochrome C oxidase deficiency in Saguenay-Lac-Saint-Jean. Am. J. Hum. Genet. 1993;53:488–496. https://pmc.ncbi.nlm.nih.gov/articles/instance/1682365/pdf/ajhg00053-0194.pdf[↩]
- Distelmaier F, Haak TB, Wortmann SB, Mayr JA, Prikisch H. Treatable mitochondrial diseases:cofactor metabolism and beyond. Brain. 2017;140(2):e11. doi: 10.1093/brain/aww303[↩][↩][↩][↩]
- Brown G. Defects of thiamine transport and metabolism. J Inherit Metab Dis. 2014;37(4):577–585. doi: 10.1007/s10545-014-9712-9[↩]
- Hargreaves IP. Coenzyme Q10 as a therapy for mitochondrial disease. Int J Biochem Cell Biol. 2014;49:105–111. doi: 10.1016/j.biocel.2014.01.020[↩]
- Tiet M.Y., Lin Z., Gao F., Jennings M.J., Horvath R. Targeted Therapies for Leigh Syndrome: Systematic Review and Steps Towards a ‘Treatabolome’. J. Neuromuscul. Dis. 2021;8:885–897. doi: 10.3233/JND-210715[↩]
- Montini G., Malaventura C., Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N. Engl. J. Med. 2008;358:2849–2850. doi: 10.1056/NEJMc0800582[↩]
- Chen Z., Zhao Z., Ye Q., Chen Y., Pan X., Sun B., Huang H., Zheng A. Mild clinical manifestation and unusual recovery upon coenzyme Q10 treatment in the first Chinese Leigh syndrome pedigree with mutation m.10197 G > A. Mol. Med. Rep. 2015;11:1956–1962. doi: 10.3892/mmr.2014.2911[↩][↩][↩][↩]
- Scalais E., Chafai R., Van Coster R., Bindl L., Nuttin C., Panagiotaraki C., Seneca S., Lissens W., Ribes A., Geers C., et al. Early myoclonic epilepsy, hypertrophic cardiomyopathy and subsequently a nephrotic syndrome in a patient with CoQ10 deficiency caused by mutations in para-hydroxybenzoate-polyprenyl transferase (COQ2) Eur. J. Paediatr. Neurol. 2013;17:625–630. doi: 10.1016/j.ejpn.2013.05.013[↩]
- Haginoya K., Miyabayashi S., Kikuchi M., Kojima A., Yamamoto K., Omura K., Uematsu M., Hino-Fukuyo N., Tanaka S., Tsuchiya S. Efficacy of idebenone for respiratory failure in a patient with Leigh syndrome: A long-term follow-up study. J. Neurol. Sci. 2009;278:112–114. doi: 10.1016/j.jns.2008.11.008[↩]
- Enns G.M., Kinsman S.L., Perlman S.L., Spicer K.M., Abdenur J.E., Cohen B.H., Amagata A., Barnes A., Kheifets V., Shrader W.D., et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol. Genet. Metab. 2012;105:91–102. doi: 10.1016/j.ymgme.2011.10.009[↩]
- Martinelli D., Catteruccia M., Piemonte F., Pastore A., Tozzi G., Dionisi-Vici C., Pontrelli G., Corsetti T., Livadiotti S., Kheifets V., et al. EPI-743 reverses the progression of the pediatric mitochondrial disease—Genetically defined Leigh Syndrome. Mol. Genet. Metab. 2012;107:383–388. doi: 10.1016/j.ymgme.2012.09.007[↩]
- Fujii T., Ito M., Miyajima T., Okuno T. Dichloroacetate therapy in Leigh syndrome with a mitochondrial T8993C mutation. Pediatr. Neurol. 2002;27:58–61. doi: 10.1016/S0887-8994(02)00378-8[↩]
- Kimura S., Osaka H., Saitou K., Ohtuki N., Kobayashi T., Nezu A. Improvement of lesions shown on MRI and CT scan by administration of dichloroacetate in patients with Leigh syndrome. J. Neurol. Sci. 1995;134:103–107. doi: 10.1016/0022-510X(95)00218-8[↩][↩]
- Spruijt L., Naviaux R.K., McGowan K.A., Nyhan W.L., Sheean G., Haas R.H., Barshop B.A. Nerve conduction changes in patients with mitochondrial diseases treated with dichloroacetate. Muscle Nerve. 2001;24:916–924. doi: 10.1002/mus.1089[↩]
- Koga Y., Povalko N., Katayama K., Kakimoto N., Matsuishi T., Naito E., Tanaka M. Beneficial effect of pyruvate therapy on Leigh syndrome due to a novel mutation in PDH E1α gene. Brain Dev. 2012;34:87–91. doi: 10.1016/j.braindev.2011.03.003[↩]
- Tanaka M., Nishigaki Y., Fuku N., Ibi T., Sahashi K., Koga Y. Therapeutic potential of pyruvate therapy for mitochondrial diseases. Mitochondrion. 2007;7:399–401. doi: 10.1016/j.mito.2007.07.002[↩]
- Komaki H., Nishigaki Y., Fuku N., Hosoya H., Murayama K., Ohtake A., Goto Y., Wakamoto H., Koga Y., Tanaka M. Pyruvate therapy for Leigh syndrome due to cytochrome c oxidase deficiency. Biochim. Biophys. Acta. 2010;1800:313–315. doi: 10.1016/j.bbagen.2009.07.008[↩]
- Fujii T., Nozaki F., Saito K., Hayashi A., Nishigaki Y., Murayama K., Tanaka M., Koga Y., Hiejima I., Kumada T. Efficacy of pyruvate therapy in patients with mitochondrial disease: A semi-quantitative clinical evaluation study. Mol. Genet. Metab. 2014;112:133–138. doi: 10.1016/j.ymgme.2014.04.008[↩][↩]
- Koene S., Spaans E., Van Bortel L., Van Lancker G., Delafontaine B., Badilini F., Beyrath J., Smeitink J. KH176 under development for rare mitochondrial disease: A first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J. Rare Dis. 2017;12:163. doi: 10.1186/s13023-017-0715-0[↩]
- A Dose-escalating Clinical Trial With KH176. https://clinicaltrials.gov/study/NCT02544217[↩]
- Janssen M.C.H., Koene S., de Laat P., Hemelaar P., Pickkers P., Spaans E., Beukema R., Beyrath J., Groothuis J., Verhaak C., et al. The KHENERGY Study: Safety and Efficacy of KH176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin. Pharmacol. Ther. 2019;105:101–111. doi: 10.1002/cpt.1197[↩]
- Garone C., Donati M.A., Sacchini M., Garcia-Diaz B., Bruno C., Calvo S., Mootha V.K., Dimauro S. Mitochondrial encephalomyopathy due to a novel mutation in ACAD9. JAMA Neurol. 2013;70:1177–1179. doi: 10.1001/jamaneurol.2013.3197[↩][↩][↩]
- Gerards M., van den Bosch B.J., Danhauser K., Serre V., van Weeghel M., Wanders R.J., Nicolaes G.A., Sluiter W., Schoonderwoerd K., Scholte H.R., et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: New function for an old gene. Brain. 2011;134:210–219. doi: 10.1093/brain/awq273[↩][↩]
- Ortigoza-Escobar J.D., Molero-Luis M., Arias A., Oyarzabal A., Darín N., Serrano M., Garcia-Cazorla A., Tondo M., Hernández M., Garcia-Villoria J., et al. Free-thiamine is a potential biomarker of thiamine transporter-2 deficiency: A treatable cause of Leigh syndrome. Brain. 2016;139:31–38. doi: 10.1093/brain/awv342[↩]
- Haack T.B., Klee D., Strom T.M., Mayatepek E., Meitinger T., Prokisch H., Distelmaier F. Infantile Leigh-like syndrome caused by SLC19A3 mutations is a treatable disease. Brain. 2014;137:e295. doi: 10.1093/brain/awu128[↩]
- Debs R., Depienne C., Rastetter A., Bellanger A., Degos B., Galanaud D., Keren B., Lyon-Caen O., Brice A., Sedel F. Biotin-responsive basal ganglia disease in ethnic Europeans with novel SLC19A3 mutations. Arch. Neurol. 2010;67:126–130. doi: 10.1001/archneurol.2009.293[↩][↩]
- Tabarki B., Alfadhel M., AlShahwan S., Hundallah K., AlShafi S., AlHashem A. Treatment of biotin-responsive basal ganglia disease: Open comparative study between the combination of biotin plus thiamine versus thiamine alone. Eur. J. Paediatr. Neurol. 2015;19:547–552. doi: 10.1016/j.ejpn.2015.05.008[↩]
- Bottani E., Lamperti C., Prigione A., Tiranti V., Persico N., Brunetti D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics. 2020;12:1083. doi: 10.3390/pharmaceutics12111083[↩]
- Viscomi C., Burlina A.B., Dweikat I., Savoiardo M., Lamperti C., Hildebrandt T., Tiranti V., Zeviani M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010;16:869–871. doi: 10.1038/nm.2188[↩][↩]
- Shayota B.J., Soler-Alfonso C., Bekheirnia M.R., Mizerik E., Boyer S.W., Xiao R., Yang Y., Elsea S.H., Scaglia F. Case report and novel treatment of an autosomal recessive Leigh syndrome caused by short-chain enoyl-CoA hydratase deficiency. Am. J. Med. Genet. A. 2019;179:803–807. doi: 10.1002/ajmg.a.61074[↩][↩]
- Tarnopolsky M.A. The mitochondrial cocktail: Rationale for combined nutraceutical therapy in mitochondrial cytopathies. Adv. Drug Deliv. Rev. 2008;60:1561–1567. doi: 10.1016/j.addr.2008.05.001[↩][↩]
- Tóth G., Morava E., Bene J., Selhorst J.J., Overmars H., Vreken P., Molnár J., Farkas V., Melegh B. Carnitine-responsive carnitine insufficiency in a case of mtDNA 8993T > C mutation associated Leigh syndrome. J. Inherit. Metab. Dis. 2001;24:421–422. doi: 10.1023/A:1010537527291[↩][↩]
- Wexler I.D., Hemalatha S.G., McConnell J., Buist N.R., Dahl H.H., Berry S.A., Cederbaum S.D., Patel M.S., Kerr D.S. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology. 1997;49:1655–1661. doi: 10.1212/WNL.49.6.1655[↩]
- Laugel V., This-Bernd V., Cormier-Daire V., Speeg-Schatz C., de Saint-Martin A., Fischbach M. Early-onset ophthalmoplegia in Leigh-like syndrome due to NDUFV1 mutations. Pediatr. Neurol. 2007;36:54–57. doi: 10.1016/j.pediatrneurol.2006.08.007[↩]
- Banka S., de Goede C., Yue W.W., Morris A.A., von Bremen B., Chandler K.E., Feichtinger R.G., Hart C., Khan N., Lunzer V., et al. Expanding the clinical and molecular spectrum of thiamine pyrophosphokinase deficiency: A treatable neurological disorder caused by TPK1 mutations. Mol. Genet. Metab. 2014;113:301–306. doi: 10.1016/j.ymgme.2014.09.010[↩][↩]
- Arii J, Tanabe Y. Leigh syndrome: serial MR imaging and clinical follow-up. AJNR Am J Neuroradiol. 2000 Sep;21(8):1502-9. https://pmc.ncbi.nlm.nih.gov/articles/PMC7974045[↩]
- Ardissone A, Bruno C, Diodato D et al. Clinical, Imaging, Biochemical and Molecular Features in Leigh Syndrome: A Study from the Italian Network of Mitochondrial Diseases. Orphanet J Rare Dis. 2021;16(1):413. doi:10.1186/s13023-021-02029-3[↩]
- Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir L, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis. 2014;9:52. doi: 10.1186/1750-1172-9-52[↩]
- Hanaford A.R., Cho Y.J., Nakai H. AAV-vector based gene therapy for mitochondrial disease: Progress and future perspectives. Orphanet J. Rare Dis. 2022;17:217. doi: 10.1186/s13023-022-02324-7[↩]
- Nakai R., Varnum S., Field R.L., Shi H., Giwa R., Jia W., Krysa S.J., Cohen E.F., Borcherding N., Saneto R.P., et al. Mitochondria transfer-based therapies reduce the morbidity and mortality of Leigh syndrome. Nat. Metab. 2024;6:1886–1896. doi: 10.1038/s42255-024-01125-5[↩]




{kind=link}