Contents
- What is Porphyria
- Porphyria common triggers
- Porphyria types
- Porphyria Cutanea Tarda
- Hepatoerythropoietic Porphyria
- Hepatoerythropoietic porphyria cause
- Hepatoerythropoietic porphyria inheritance pattern
- Hepatoerythropoietic porphyria pathophysiology
- Hepatoerythropoietic porphyria sgns and symptoms
- Hepatoerythropoietic porphyria diagnosis
- Hepatoerythropoietic porphyria differential diagnosis
- Hepatoerythropoietic porphyria treatment
- Hepatoerythropoietic Porphyria Prognosis
- Erythropoietic protoporphyria (Protoporphyria)
- X-Linked Protoporphyria
- Congenital erythropoietic porphyria
- Acute intermittent porphyria
- Acute intermittent porphyria cause
- Acute intermittent porphyria inheritance pattern
- Acute intermittent porphyria pathophysiology
- Acute intermittent porphyria types
- Acute intermittent porphyria signs and symptoms
- Acute intermittent porphyria complications
- Acute intermittent porphyria diagnosis
- Acute intermittent porphyria differential diagnosis
- Acute intermittent porphyria treatment
- Acute intermittent porphyria prognosis
- Variegate porphyria
- Hereditary coproporphyria
- ALA dehydratase deficiency porphyria
- Porphyria complications
- Porphyria causes
- Porphyria symptoms
- Porphyria diagnosis
- Porphyria treatment
What is Porphyria
Porphyria is the umbrella term for a group of rare inherited disorders or passed down from parents to children caused by abnormalities in the chemical steps that lead to ‘heme’ or ‘haem’ production. Heme (haem) is a vital molecule for all of your body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme is a component of several iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme (haem) is also found in myoglobin, a protein found in certain muscles. Normally, your body makes heme in a multi-step process (see Figures 5 and 6 below). Porphyrins are made during several steps of this process. People with porphyria are lacking certain enzymes needed for this process. This causes abnormal amounts of porphyrins or related chemicals to build up in your body. Porphyria occurs when the body cannot convert naturally occurring compounds called ‘porphyrins’ into heme (haem). Porphyrins are substances that are required for the production of red blood cells. A common feature in all porphyrias is the accumulation in the body of porphyrins or porphyrin precursors. Although these are normal body chemicals, they normally do not accumulate. Precisely which of these chemicals builds up depends on the type of porphyria. Drugs, infection, alcohol, and hormones such as estrogen may trigger attacks of certain types of porphyria.
Researchers have identified at least 8 types of porphyria, which are distinguished by their genetic cause and their signs and symptoms and are all caused by a build up of porphyrins in the cells of the body. People who have porphyria can experience a wide range of symptoms depending on the type of porphyria they have. There are 2 main types of porphyrias. One affects the skin (cutaneous porphyrias) and the other affects the nervous system (acute porphyrias). Some types of porphyria, called cutaneous porphyrias, primarily affect the skin. The most common type is porphyria cutanea tarda (PCT), which affects about 5 to 10 out of every 100,000 people 1. Areas of skin exposed to the sun become fragile and blistered, which can lead to infection, scarring, changes in skin coloring (pigmentation), and increased hair growth. Cutaneous porphyrias include congenital erythropoietic porphyria (CEP), erythropoietic protoporphyria (EPP), hepatoerythropoietic porphyria (HEP), and porphyria cutanea tarda (PCT). The most common type of porphyria in children is a cutaneous porphyria called erythropoietic protoporphyria (EPP) 2.
Symptoms of cutaneous porphyrias include:
- oversensitivity to sunlight
- blisters on exposed areas of the skin
- itching and swelling on exposed areas of the skin
The nervous system type of porphyria is called acute porphyria. Symptoms include pain in the chest, abdomen, limbs, or back; muscle numbness, tingling, paralysis, or cramping; vomiting; constipation; and personality changes or mental disorders. These symptoms come and go. Acute porphyrias are described as “acute” because their signs and symptoms appear quickly and usually last a short time. Episodes of acute porphyria can cause abdominal pain, vomiting, constipation, and diarrhea. During an episode, a person may also experience muscle weakness, seizures, fever, and mental changes such as anxiety and hallucinations. These signs and symptoms can be life-threatening, especially if the muscles that control breathing become paralyzed. Acute porphyrias include acute intermittent porphyria (AIP) and ALA dehydratase deficiency porphyria. The most common type of acute porphyria is acute intermittent porphyria (AIP). Two other forms of acute porphyria, hereditary coproporphyria (HCP) and variegate porphyria (VP), can have both acute (nervous system) and cutaneous (skin) symptoms (see Table 1 below).
Certain triggers can cause an attack, including some medicines, smoking, drinking alcohol, infections, stress, and sun exposure. Attacks develop over hours or days. They can last for days or weeks.
Symptoms of acute porphyrias include:
- pain in the abdomen—the area between the chest and hips
- pain in the chest, limbs, or back
- nausea and vomiting
- constipation—a condition in which an adult has fewer than three bowel movements a week or a child has fewer than two bowel movements a week,
- depending on the person
- urinary retention—the inability to empty the bladder completely
- confusion
- hallucinations
- seizures and muscle weakness
Symptoms of acute porphyrias can develop over hours or days and last for days or weeks. These symptoms can come and go over time, while symptoms of cutaneous porphyrias tend to be more continuous. Porphyria symptoms can vary widely in severity.
Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of the disorder. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
The porphyrias are rare diseases. Taken together, all forms of porphyria afflict fewer than 200,000 people in the United States 3. Based on European studies, the prevalence of the most common porphyria, porphyria cutanea tarda (PCT) is 1 in 10,000 and the most common acute porphyria, acute intermittent porphyria (AIP) is about 1 in 20,000, and the most common erythropoietic porphyria, erythropoietic protoporphyria (EPP), is estimated at 1 in 50,000 to 75,000 4. Congenital erythropoietic porphyria (CEP) is extremely rare with prevalence estimates of 1 in 1,000,000 or less. Only 6 cases of ALA dehydratase-deficiency porphyria (ADP) are documented 4.
Acute porphyria is more common in females than in males and often begins when people are between the ages of 15 and 45 5. Among types of cutaneous porphyria, porphyria cutanea tarda most often develops in people older than age 40, usually men 1. For other types of cutaneous porphyria, symptoms often appear in early childhood.
The porphyrias can also be split into erythropoietic (red blood cell) and hepatic (liver) types, depending on where damaging compounds called porphyrins and porphyrin precursors first build up in the body. In erythropoietic porphyrias, these compounds originate in the bone marrow. Erythropoietic porphyrias include erythropoietic protoporphyria (EPP) and congenital erythropoietic porphyria (CEP). Health problems associated with erythropoietic porphyrias include a low number of red blood cells (anemia) and enlargement of the spleen (splenomegaly). The other types of porphyrias are considered hepatic porphyrias. In these disorders, porphyrins and porphyrin precursors originate primarily in the liver, leading to abnormal liver function and an increased risk of developing liver cancer.
Figure 1. Porphyrin molecular structure
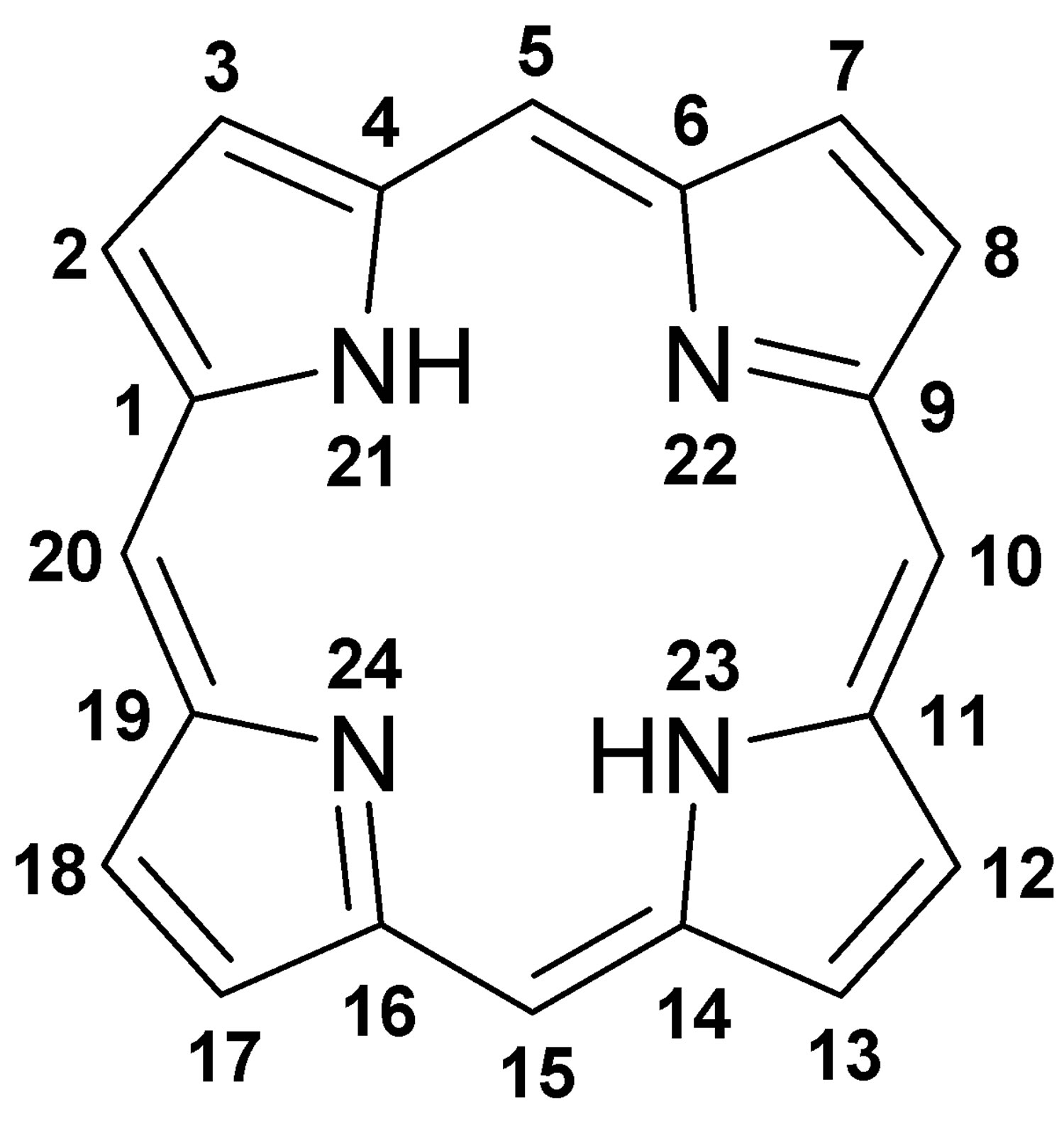
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 6 ]Figure 2. Hemoglobin molecular structure
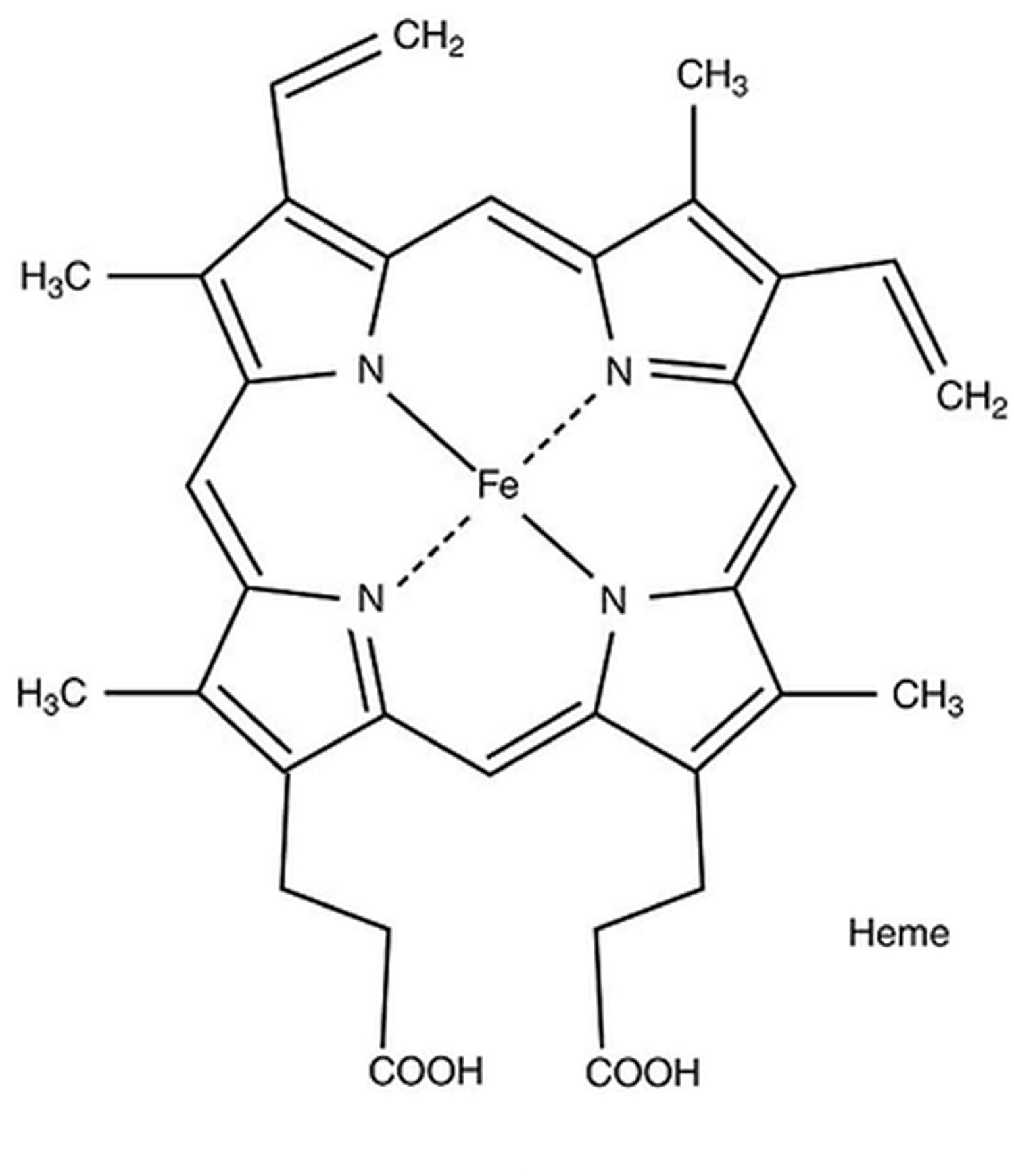
Figure 3. Heme (haem) – oxygenation of heme protein
Figure 4. Heme (haem) molecular structure
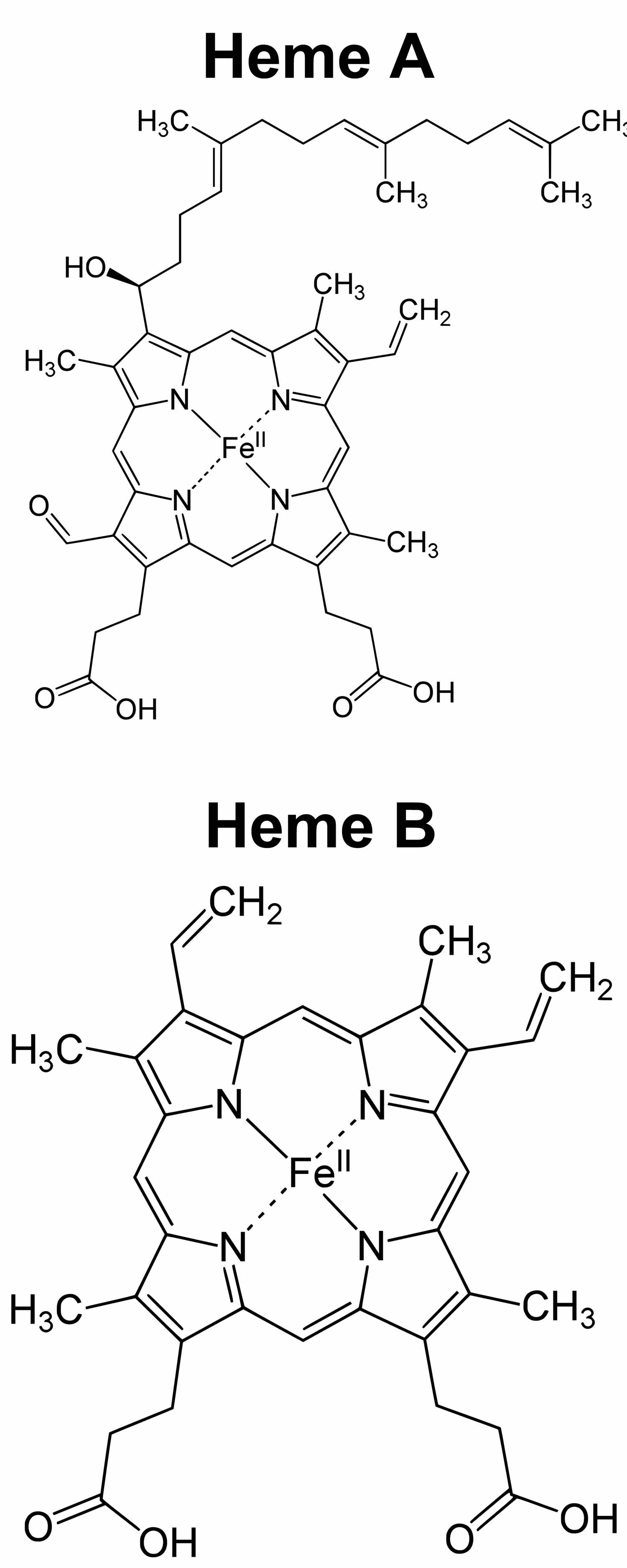
Footnote: Heme A and heme B molecular structures
[Source 8 ]Figure 5. Heme biosynthesis pathway
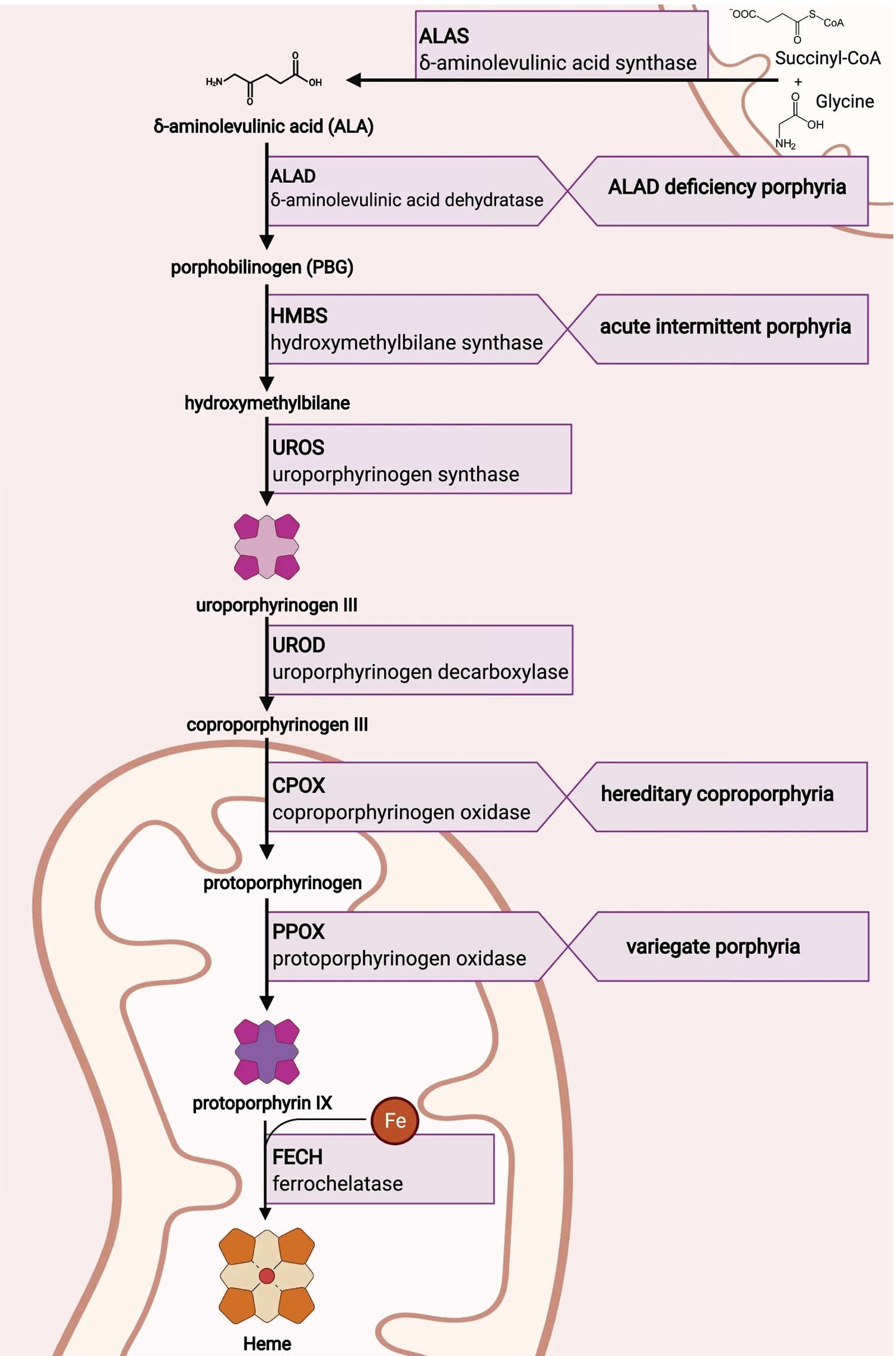
Heme synthesis
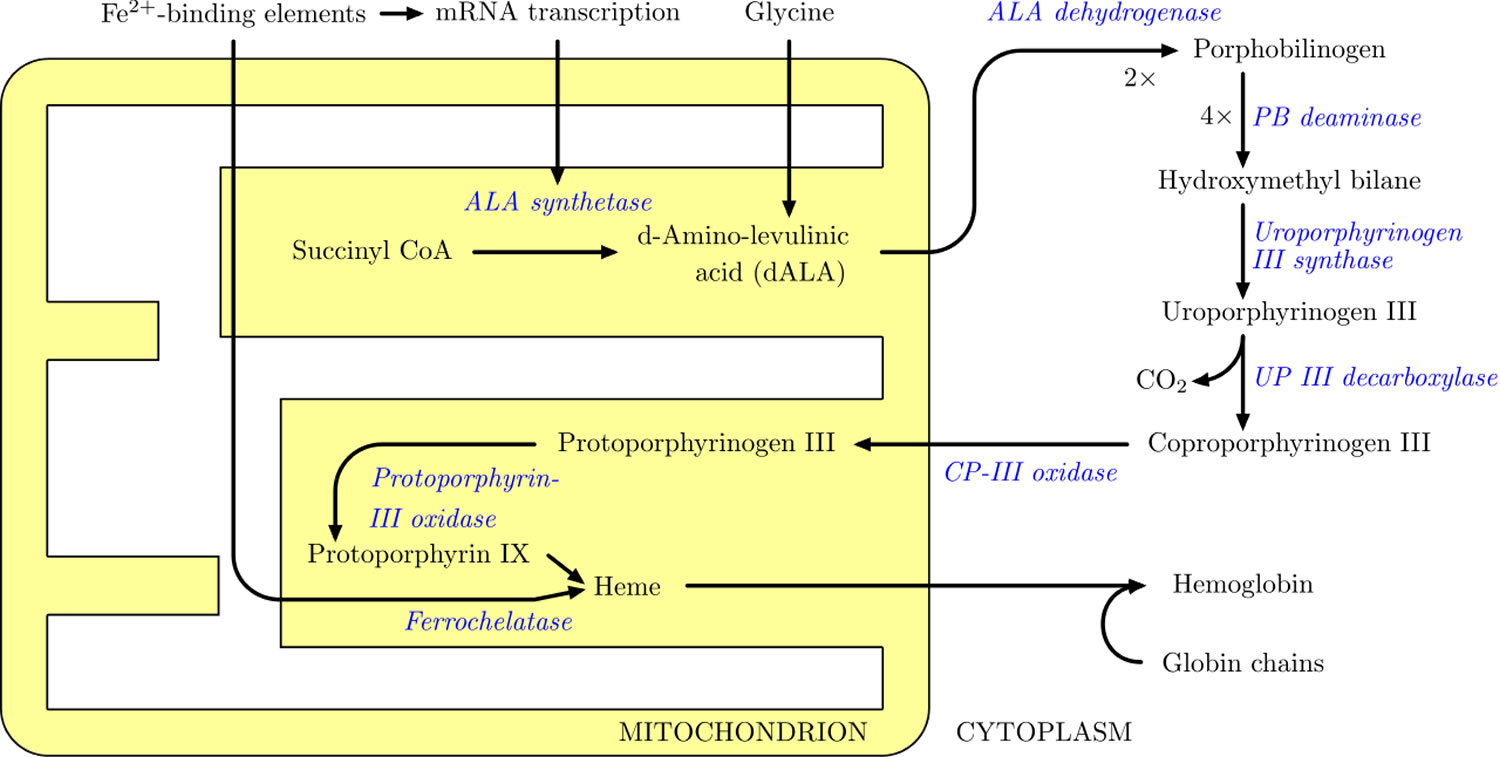
Figure 6. Heme synthesis pathway
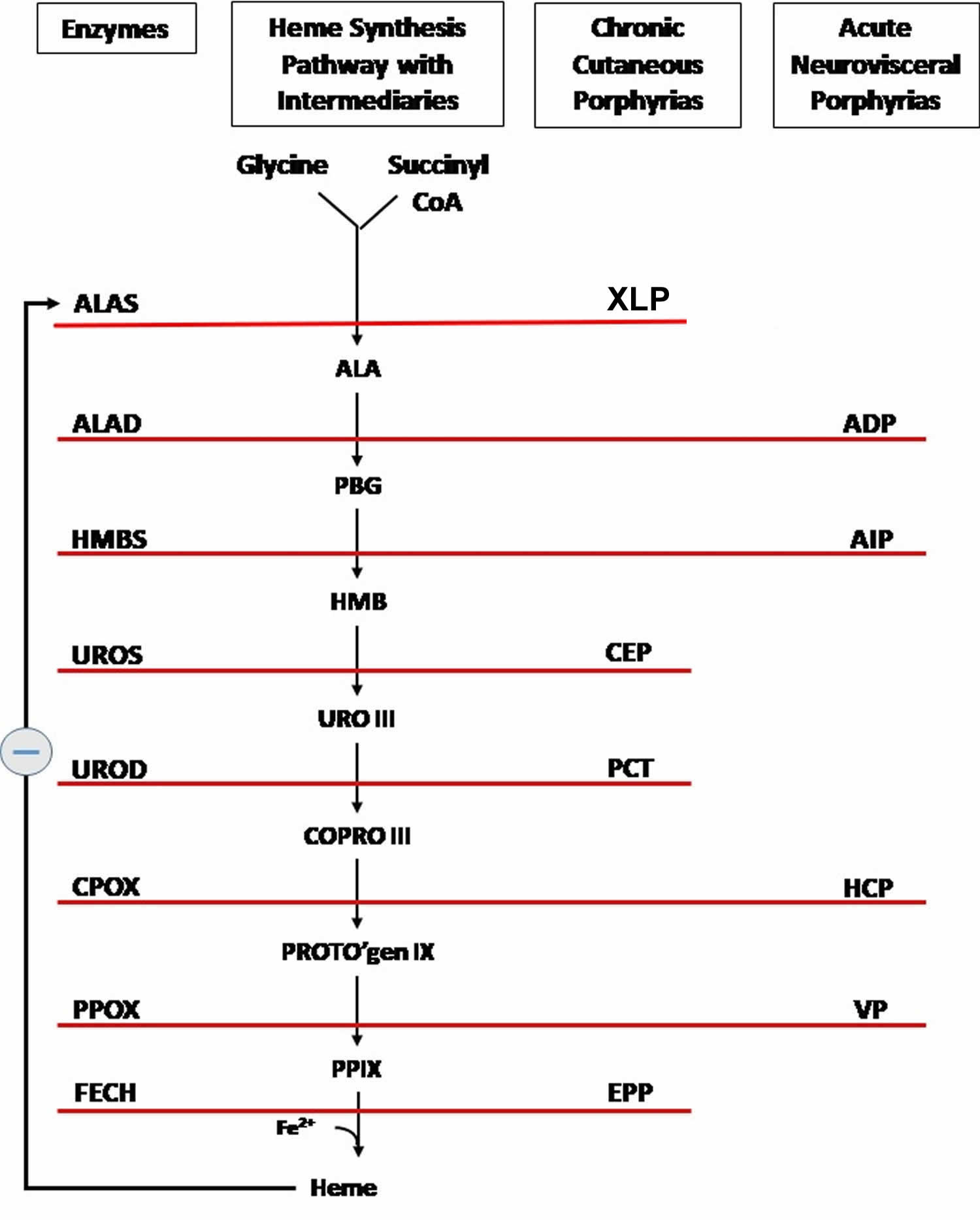
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 10 ]Porphyria common triggers
Various triggers can prompt the development of porphyria. Environmental factors can strongly influence the occurrence and severity of signs and symptoms of porphyria. Alcohol, smoking, certain drugs, hormones, other illnesses, stress, and dieting or periods without food (fasting) can all trigger the signs and symptoms of some forms of porphyria. Additionally, exposure to sunlight worsens the skin damage in people with cutaneous porphyrias.
While the factors in the following list may seem to have nothing in common, each one demands increased heme production, which overwhelms the body’s ability to deal with the increased levels of porphyrins.
Common triggers include:
- Prescription drugs such as barbiturates, tranquilizers, sedatives, oral contraceptives and some types of antibiotics
- Female sex hormones
- Sunlight
- Alcohol
- Cigarette smoking
- Infection
- Surgery
- Fasting.
In most cases, the cause is a combination of genetic and environmental factors. More women than men are affected for reasons unknown. There is no cure but treatments are available to manage the symptoms.
Porphyria can be hard to diagnose. It requires blood, urine, and stool tests. Each type of porphyria is treated differently. Treatment may involve avoiding triggers, receiving heme through a vein, taking medicines to relieve symptoms, or having blood drawn to reduce iron in the body. People who have severe attacks may need to be hospitalized.
Porphyria types
There are at least 8 types of porphyria, with the two most common being:
- Cutaneous porphyrias. All but one of the cutaneous porphyrias cause painful skin blistering and fragility on sun-exposed areas of the body, most commonly the backs of the hands, forearms, face, ears and neck (photosensitivity):
- The cutaneous porphyrias are sub-categorized as:
- Porphyria cutanea tarda (PCT)
- Hepatoerythropoietic Porphyria (HEP)
- Erythropoietic protoporphyria (EPP)
- Congenital erythropoietic porphyria (CEP)
- X-Linked Protoporphyria (XLP)
- Congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP) occur in childhood with severe blistering skin lesions. Porphyria cutanea tarda (PCT) occurs in adulthood generally and has less severe blistering skin lesions after sun exposure. Erythropoietic Protoporphyria (EPP) and X-linked Protoporphyria (XLP) have the same symptoms of painful, but non-blistering, reactions to sunlight. There can also be swelling and redness of the sun exposed areas of the skin with Erythropoietic Protoporphyria (EPP) and X-linked Protoporphyria (XLP).
- The cutaneous porphyrias are sub-categorized as:
- Acute porphyrias, mainly affecting the neurological system characterized by intense pain, confusion and limb weakness:
- The acute porphyrias are sub-categorized as:
- Acute intermittent porphyria (AIP)
- Hereditary coproporphyria (HCP)
- Variegate porphyria (VP)
- ALA dehydratase deficiency porphyria (ADP)
- The acute porphyrias are sub-categorized as:
Porphyria is usually inherited, but it can also occur without anyone else in the family having it.
Experts also classify porphyrias as erythropoietic (red blood cell) or hepatic (liver):
- In erythropoietic porphyrias, the body overproduces porphyrins, mainly in the bone marrow.
- In hepatic porphyrias, the body overproduces porphyrins and porphyrin precursors, mainly in the liver.
Table 1 lists each type of porphyria, the deficient enzyme responsible for the disorder, and the main location of porphyrin buildup.
Table 1. Types of porphyria
| Type of Porphyria | Deficient Enzyme | Main Location of Porphyrin Buildup | Parts of the Body Affected |
|---|---|---|---|
| ALA-dehydratase deficiency porphyria | delta-aminolevulinic acid dehydratase | Liver | Nervous system |
| Acute intermittent porphyria | porphobilinogen deaminase | Liver | Nervous system |
| Hereditary coproporphyria | coproporphyrinogen oxidase | Liver | Nervous system and skin |
| Variegate porphyria | protoporphyrinogen oxidase | Liver | Nervous system and skin |
| Congenital erythropoietic porphyria | uroporphyrinogen III cosynthase | Bone marrow | Skin |
| Porphyria cutanea tarda | uroporphyrinogen decarboxylase (~75% deficiency) | Liver | Skin |
| Hepatoerythropoietic porphyria | uroporphyrinogen decarboxylase (~90% deficiency) | Bone marrow | Skin |
| Protoporphyrias: erythropoietic protoporphyria and x-linked protoporphyria | ferrochelatase (~75% deficiency) | Bone marrow | Skin |
Porphyria Cutanea Tarda
Porphyria Cutanea Tarda (PCT) is the most common of all porphyrias (one of the hepatic porphyrias) and results from a deficiency of the enzyme uroporphyrinogen decarboxylase or uroporphyrinogen III decarboxylase (UROD) 11, 12, 13, 14, 15, 16, 17. The hallmark of porphyria cutanea tarda (PCT) is photosensitivity (sunlight sensitivity or abnormal skin reaction triggered by exposure to sunlight or other forms of ultraviolet (UV) light) 18, 19. Liver (hepatic) uroporphyrinogen decarboxylase or uroporphyrinogen III decarboxylase (UROD) enzyme is tasked with the conversion of uroporphyrinogen III to coproporphyrinogen III, the fifth step in heme biosynthesis and failure to do so results in the accumulation of the preceding compounds (highly carboxylated porphyrinogens predominately uroporphyrinogen and the porphyrinogens that get oxidized to their respective porphyrins, uroporphyrin and 7-carboxylate porphyrin) in the liver that eventually appears in the plasma and urine 20, 21. The accumulation of these toxic compounds (uroporphyrin and 7-carboxylate porphyrin) in different organs, especially the liver and skin, leads to the signs and symptoms seen in porphyria cutanea tarda patients. Porphyrins are photoactive molecules that efficiently absorb light energy in the visible violet spectrum. Photoexcited porphyrins in the skin cause oxidative damage to biomolecular targets, causing the skin lesions in porphyria cutanea tarda. The most common photocutaneous manifestations of porphyria cutanea tarda are due to increased mechanical fragility after sunlight exposure; erosions and blisters form painful indolent sores that heal with milia, dyspigmentation, and scarring. An important point to note is that the uroporphyrinogen decarboxylase or uroporphyrinogen III decarboxylase (UROD) enzymatic activity should drop below 20% before signs and symptoms can be seen 11.
Porphyria cutanea tarda (PCT) is essentially an acquired disease, but some individuals have a genetic (autosomal dominant) deficiency of enzyme uroporphyrinogen decarboxylase (UROD) that contributes to development of familial porphyria cutanea tarda (F-PCT).
- Porphyria cutanea tarda type 1 (Sporadic porphyria cutanea tarda): In approximately 75% to 80% of cases the uroporphyrinogen decarboxylase (UROD) enzyme deficiency is associated with an underlying liver diseases that include iron overload from hemochromatosis, multiple blood transfusions, or iron supplements, chronic hepatitis B and hepatitis C infection and excessive alcohol consumption resulting in the inhibition of uroporphyrinogen decarboxylase (UROD) enzyme, the fifth enzyme in the heme synthetic pathway in the liver. Hormones such as oral contraceptive or hormone replacement therapy may also trigger porphyria cutanea tarda. Kidney dialysis patients can also develop porphyria cutanea tarda as they cannot excrete the porphyrins. Rarely, other conditions such as systemic lupus erythematosus (SLE) and human immunodeficiency virus (HIV) infection can cause porphyria cutanea tarda. Furthermore, many patients have more than one risk factor.
- Porphyria cutanea tarda type 2 (Familial porphyria cutanea tarda or F-PCT): In the remaining cases (20% to 25%), individuals have a genetic predisposition to developing porphyria cutanea tarda (PCT), specifically a mutation in the UROD (uroporphyrinogen decarboxylase) gene inherited from one parent and are classified as having familial porphyria cutanea tarda (F-PCT). Most individuals with UROD genetic mutation do not develop porphyria cutanea tarda (PCT); the mutation is a predisposing factor and additional factors are required for the development of the disorder in these individuals. These factors are called susceptibility factors and are required for the development of both sporadic and familial porphyria cutanea tarda.
- Porphyria cutanea tarda type 3. This rare type of porphyria cutanea tarda is very similar to type 1 porphyria cutanea tarda because of normal UROD genes 22. Yet, type 3 porphyria cutanea tarda is observed in more than one family member suggesting the presence of a genetic mechanism other than UROD gene mutation 23.
- In extremely rare cases, individuals have mutations in both UROD (uroporphyrinogen decarboxylase) genes. This autosomal recessive form of familial porphyria cutanea tarda or Homozygous familial porphyria cutanea tarda is known as hepatoerythropoietic porphyria (HEP). Hepatoerythropoietic porphyria (HEP) occurs in childhood and is usually more severe than porphyria cutanea tarda types 1 sporadic or 2 familial porphyria cutanea tarda 24. Mild cases of hepatoerythropoietic porphyria (HEP) may resemble porphyria cutanea tarda (PCT) but are readily differentiated by marked elevation in erythrocyte zinc protoporphyrin.
Generally, porphyria cutanea tarda develops in mid to late adulthood. Porphyria cutanea tarda symptoms usually occur after the age of 30 and its onset in childhood is rare. The symptoms of porphyria cutanea tarda (PCT) are limited to your skin. It does not cause people to become acutely unwell, as in the acute types of porphyria. Sun-exposed areas of your skin most commonly the backs of your hands can become friable and prone to blistering, scarring and excess hair growth.
Porphyria cutanea tarda is found worldwide that affect both males and females equally and in individuals of all races. Porphyria cutanea tarda is a rare disorder with the prevalence being estimated to be approximately 1 in 10,000 to 25,000 individuals in the general population. Porphyria cutanea tarda (PCT) has a prevalence of about 1 in 10,000 people according to a Norwegian study and occurs most commonly in middle-aged adults 25. A 1 in 25000 prevalence has been reported in the United States 23.
Porphyria cutanea tarda (PCT) is a multifactorial disorder, which means that several different factors such as genetic and environmental factors occurring in combination are necessary for the development of porphyria cutanea tarda. These factors are not necessarily the same for each individual. These factors contribute either directly or indirectly to decreased levels or ineffectiveness of an enzyme known as uroporphyrinogen decarboxylase (UROD) within your liver. When uroporphyrinogen decarboxylase (UROD) levels in your liver decrease to approximately 20% of normal levels, the symptoms of porphyria cutanea tarda may develop.
The uroporphyrinogen decarboxylase (UROD) enzyme is essential for breaking down (metabolizing) certain chemicals in the body known as porphyrins. Low levels of functional uroporphyrinogen decarboxylase (UROD) result in the abnormal accumulation of specific porphyrins in your body, especially within the blood, liver and skin. The symptoms of porphyria cutanea tarda occur because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in your skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin (photosensitivity) found in individuals with porphyria cutanea tarda. Your liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in your liver, they can cause toxic damage to your liver.
The exact, underlying mechanisms that cause porphyria cutanea tarda are complex and varied. It is determined that iron accumulation within the liver plays a central role in the development of the disorder in most individuals (sporadic or acquired porphyria cutanea tarda [PCT type 1]). Recently, researchers have discovered that a substance called uroporphomethene, which is an oxidized form of a specific porphyrin known as uroporphyrinogen, is an inhibitor that reduces the activity of the uroporphyrinogen decarboxylase (UROD) enzyme in the liver. The oxidation of uroporphyrinogen into uroporphomethene has been shown to be iron dependent, emphasizing the importance or elevated iron levels in the development of porphyria cutanea tarda.
The relationship between iron levels and porphyria cutanea tarda has long been established and porphyria cutanea tarda is classified as an iron-dependent disease. Clinical symptoms often correlate with abnormally elevated levels of iron in the liver (iron overloading). Iron overloading in the liver may only be mild or moderate. The exact relationship between iron accumulation and porphyria cutanea tarda is not fully understood, however, as there is no specific level of iron in the liver that correlates to disease in porphyria cutanea tarda (e.g. some individuals with symptomatic porphyria cutanea tarda have normal iron levels).
Porphyria Cutanea Tarda (PCT) becomes active when predisposing factors such as excess iron, alcohol, chronic hepatitis C (hepatitis C Virus [HCV]), HIV infection, estrogens (used, for example, in oral contraceptives and prostate cancer treatment) and possibly smoking, combine to cause a deficiency of enzyme uroporphyrinogen decarboxylase (UROD) in the liver 26. Hemochromatosis, a inherited iron overload disorder, also can predispose individuals to porphyria cutanea tarda. Some of these factors and marked accumulation of porphyrins in the liver due to porphyria cutanea tarda (PCT) itself can lead to chronic liver damage and liver cancer.
The treatment of porphyria cutanea tarda is directed toward the underlying liver problem and the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, general internists, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan your treatment.
The first step in the management of porphyria cutanea tarda is the avoidance of all risk factors such as strictly avoiding alcohol, smoking, and estrogen therapy, along with limiting any excess intake of iron. Since porphyria cutanea tarda (PCT) is a photosensitive skin condition, sunlight avoidance is the key until the porphyrin levels have normalized. The wavelengths inducing porphyrins are in the range of 400-410 nm, and only titanium dioxide or zinc oxide containing sunscreen is effective 27. Protective clothing is also helpful in protecting the skin from harmful sunlight rays. Any affected skin areas should be kept clean to prevent the development of skin infections, and associated pain can be managed with oral analgesics.
Presently, there are no effective treatments that restore UROD enzyme levels in individuals with familial porphyria cutanea tarda (F-PCT) 28. However, treatment seems to be equally effective in familial porphyria cutanea tarda (F-PCT) and non-familial porphyria cutanea tarda. Factors that tend to activate the disease should be removed and may result in the resolution of porphyria cutanea tarda. Treatment may include reducing alcohol consumption, stopping estrogen or hormone treatment, avoiding excessive iron intake, or antiviral treatment for underlying hepatitis C.
Reduction of liver iron content is the general recommendation and the most widely recommended treatment is a schedule of repeated phlebotomies (removal of blood), with the aim of reducing iron in the liver 28, 23, 29, 30, 31. This actually reduces iron stores throughout the body. Usually, removal of only 5 to 6 pints of blood (one pint [approximately 450 ml of blood] every one to two weeks) is sufficient, which indicates that iron stores are not excessively increased in most porphyria cutanea tarda patients. The best guides to response are measurements of serum ferritin and plasma porphyrins. Phlebotomies are stopped when the ferritin falls to ~20 ng/ml 32, 33. Normal ferritin levels vary by gender and age, but generally, for adult males, ferritin level is between 30-300 ng/mL, and for adult females, it’s between 13-150 ng/mL.
Iron chelation therapy i.e., deferasirox or deferoxamine may be considered when phlebotomy is contraindicated, and low iron diet may be beneficial if the latter fails 28.
If phlebotomy cannot be done, as in elderly patients or those who are anemic, antimalarial tablets such as low doses of either chloroquine (125mg twice weekly) or hydroxychloroquine (100mg twice weekly) to allow the porphyrins to be excreted more easily. Usual dosages of these drugs should not be used because they can cause transient but sometimes severe liver damage and worsening of photosensitivity in porphyria cutanea tarda patients.
Furthermore, use of antiviral therapy may benefit patients with chronic hepatitis C infections and reduce risk of progressing to liver cancer 29.
After treatment for porphyria cutanea tarda, periodic measurement of plasma porphyrins may be advised, especially if a contributing factor such as estrogen exposure is resumed. If a recurrence does occur, it can be detected early and treated promptly. The treatment of porphyria cutanea tarda is almost always successful, and the prognosis is usually excellent.
Figure 7. Porphyria Cutanea Tarda
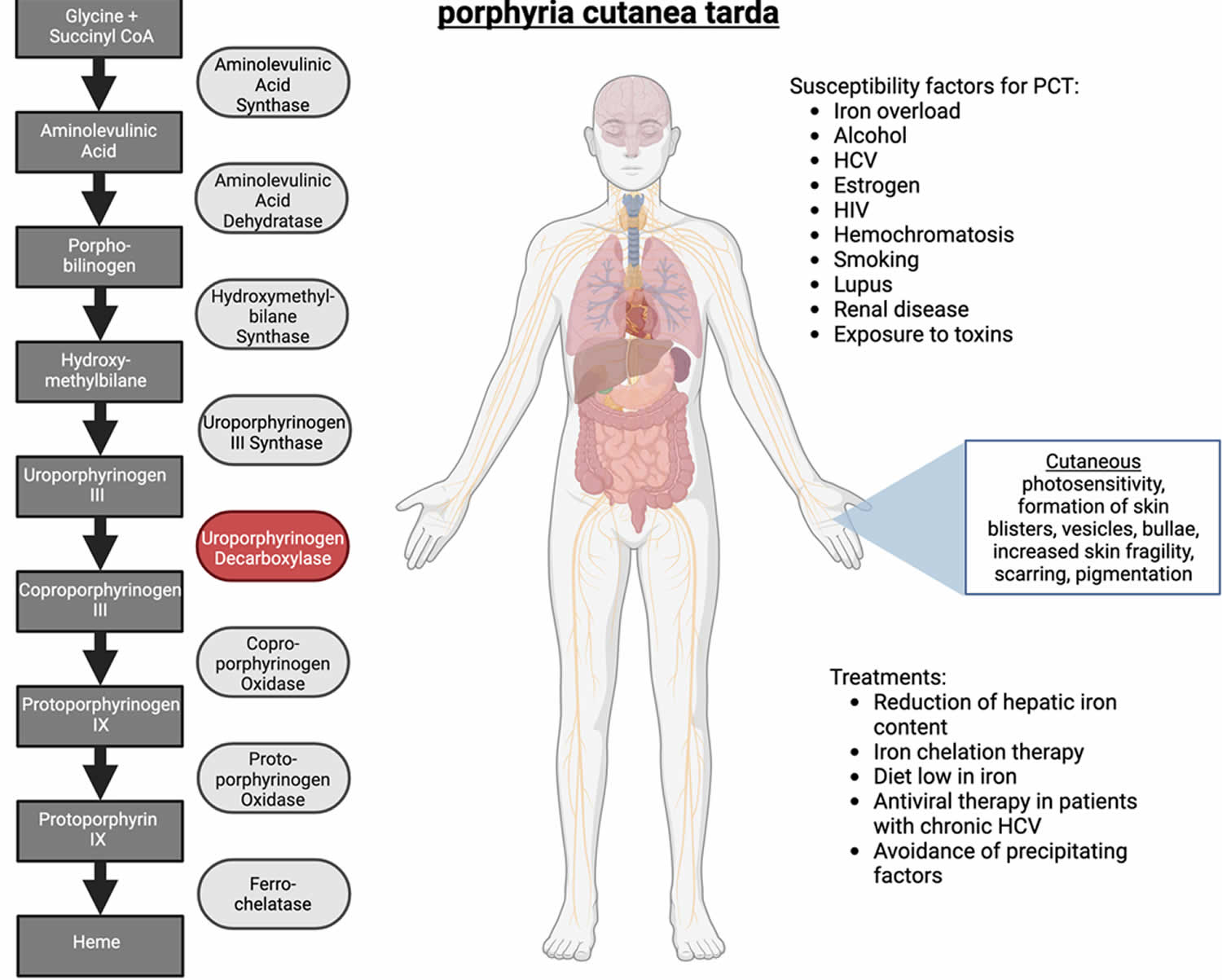
Footnotes: Porphyria cutanea tarda (PCT) is caused by deficiency in uroporphyrinogen decarboxylase (UROD) enzyme, and is subdivided into two categories: Type 1 (sporadic PCT), and Type 2 (familial PCT). Porphyria cutanea tarda (PCT) presents mainly with skin manifestations which are triggered by susceptibility factors (risk factors) such as alcohol, hepatitis C virus (HCV), estrogen, hemochromatosis, smoking, and others. Porphyria cutanea tarda (PCT) is the most common type of porphyria, and is managed by reduction of hepatic iron content and avoidance of susceptibility factors.
[Source 34 ]Figure 8. Porphyria cutanea tarda
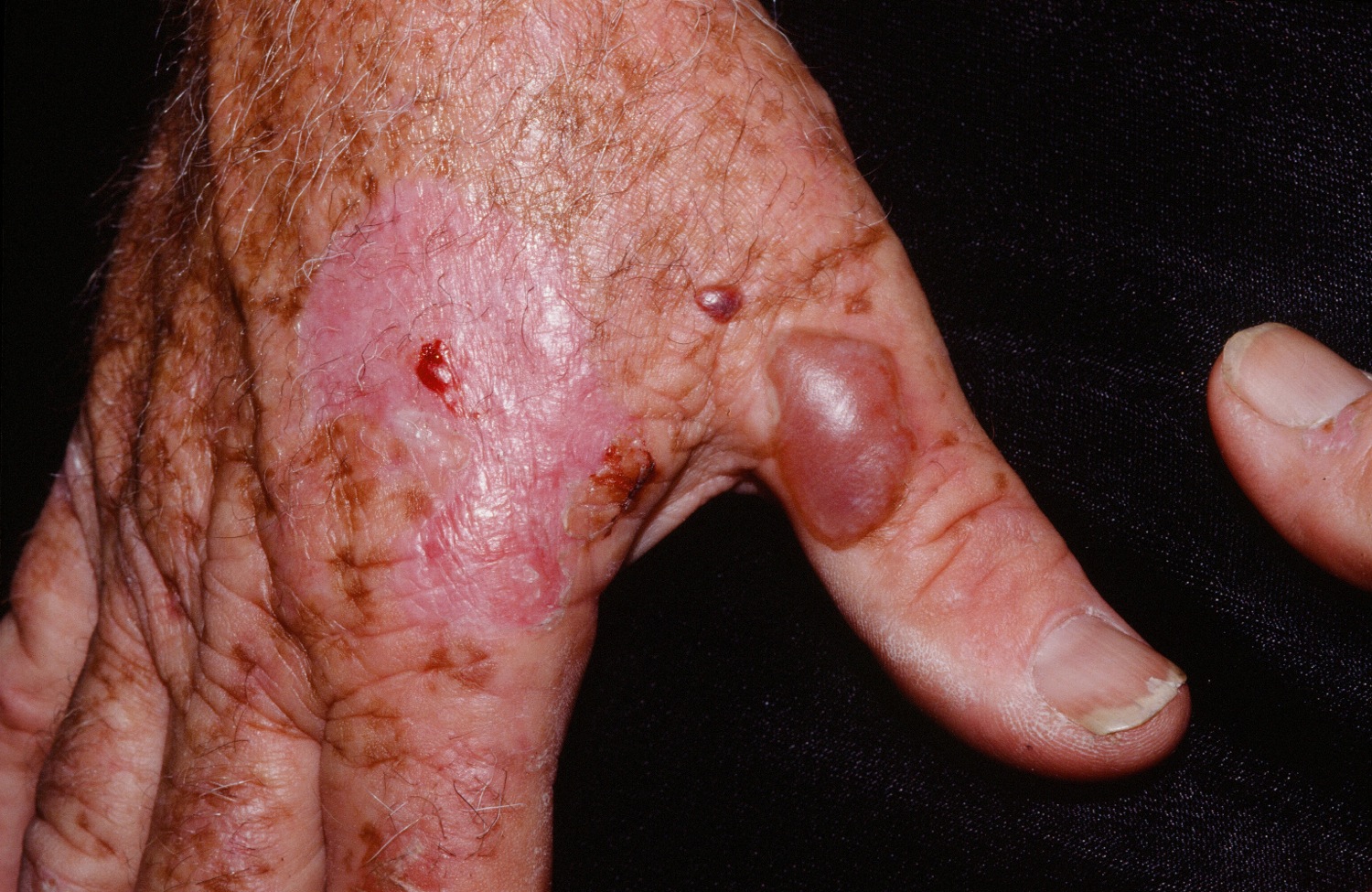
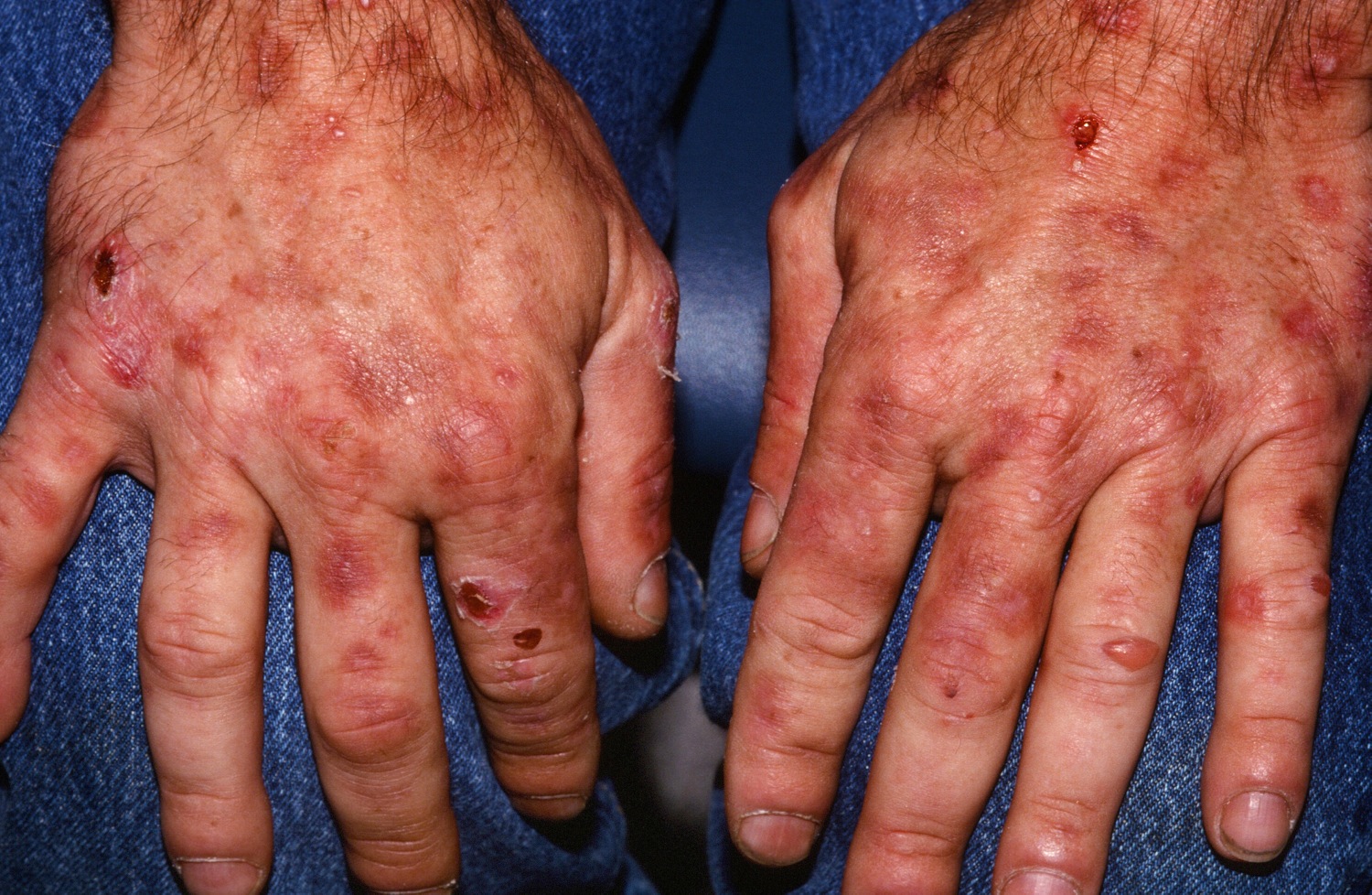
Figure 9. Porphyria cutanea tarda pathophysiology
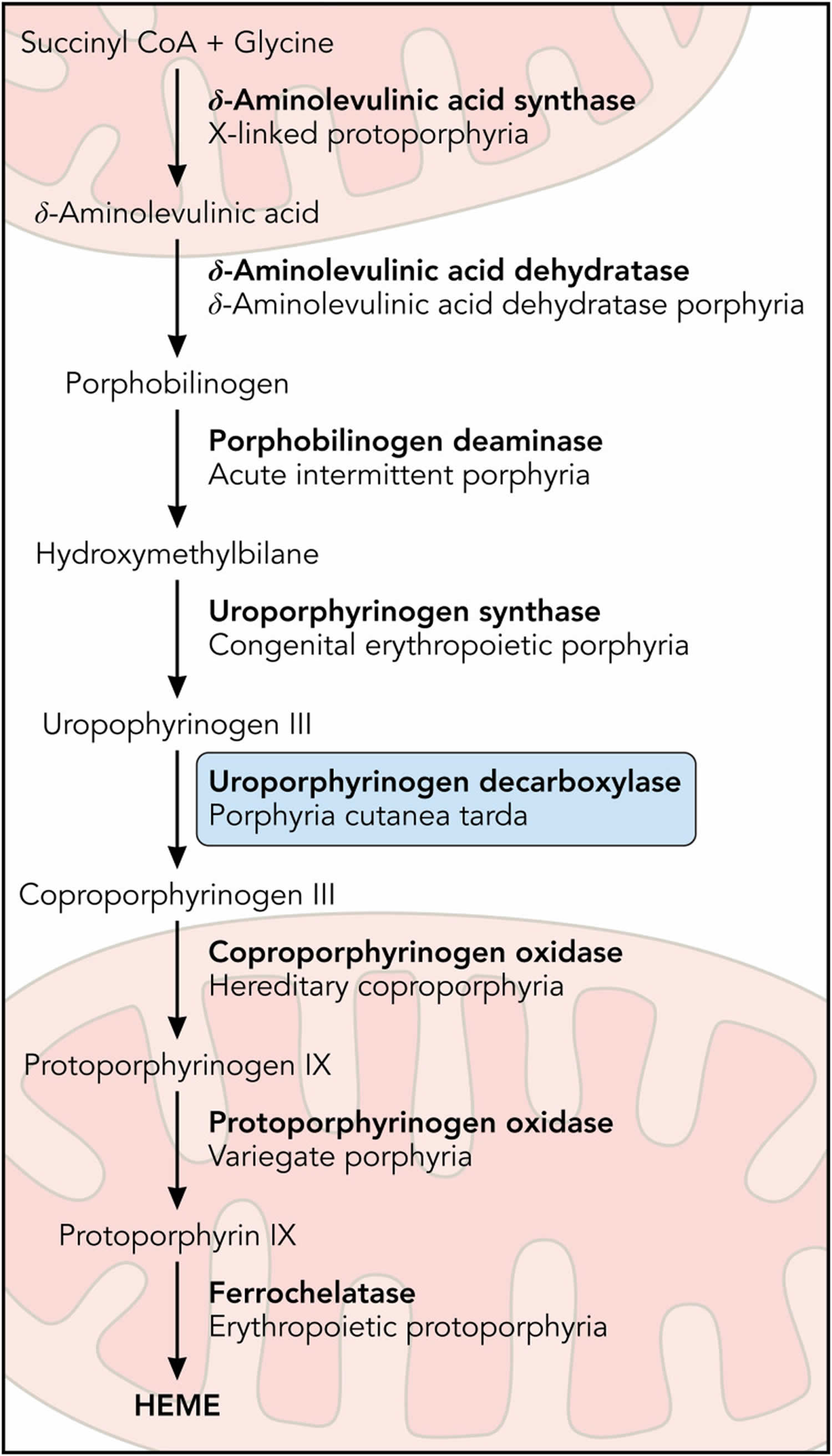
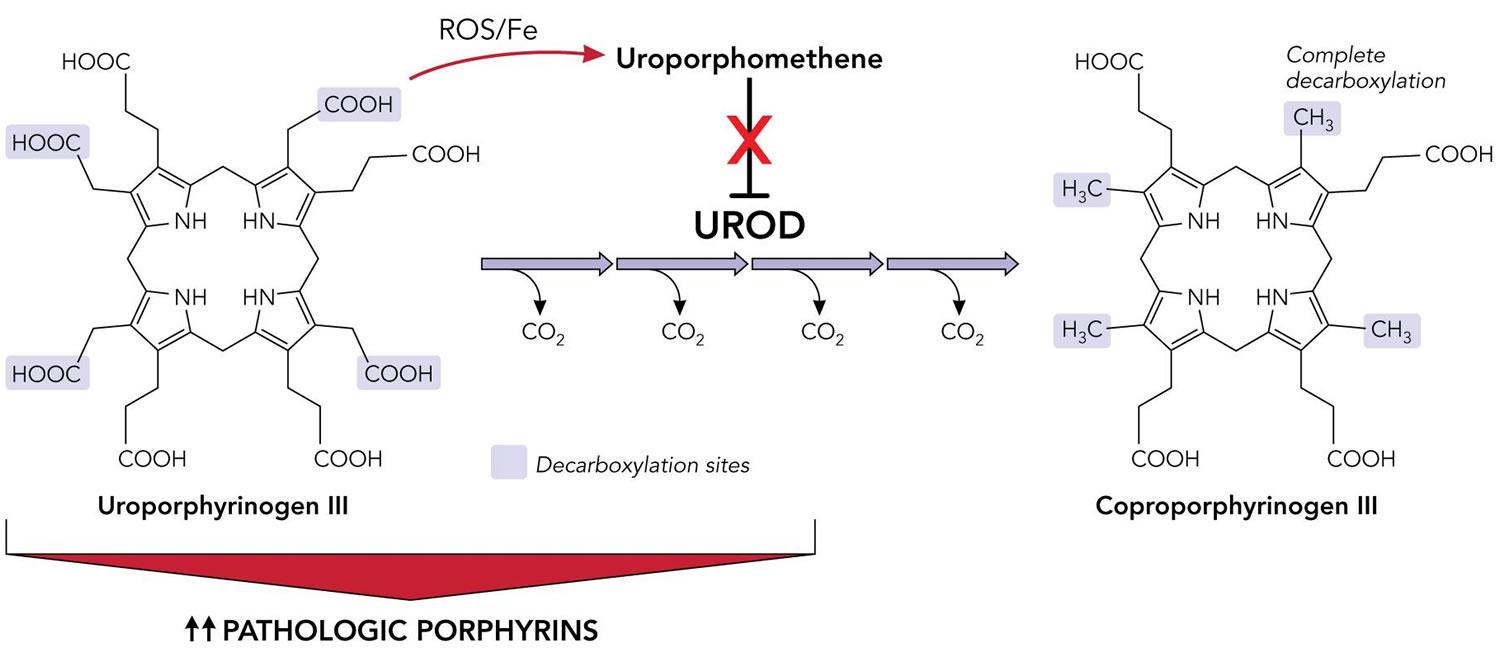
Footnotes: Inhibition of uroporphyrinogen decarboxylase (UROD) enzyme by uroporphomethene leads to the accumulation of porphyrins and manifestations of disease in porphyria cutanea tarda (PCT). Under normal conditions, UROD (uroporphyrinogen decarboxylase) converts uroporphyrinogen to coproporphyrinogen by a series of 4 sequential decarboxylations. In the presence of iron and free radicals, uroporphyrinogen is partially oxidized, leading to the formation of a uroporphomethene inhibitor of UROD (uroporphyrinogen decarboxylase). Decarboxylated uroporphyrinogen intermediates subsequently accumulate and auto-oxidize to their corresponding porphyrins, predominantly uroporphyrins. Photosensitive porphyrins accumulate in the plasma and are responsible for the cutaneous manifestations of porphyria cutanea tarda (PCT).
Abbreviations: Fe = iron; PCT = porphyria cutanea tarda; UROD = uroporphyrinogen decarboxylase; ROS = reactive oxygen species.
[Source 12 ]Figure 10. Porphyria cutanea tarda diagnostic and treatment algorithm
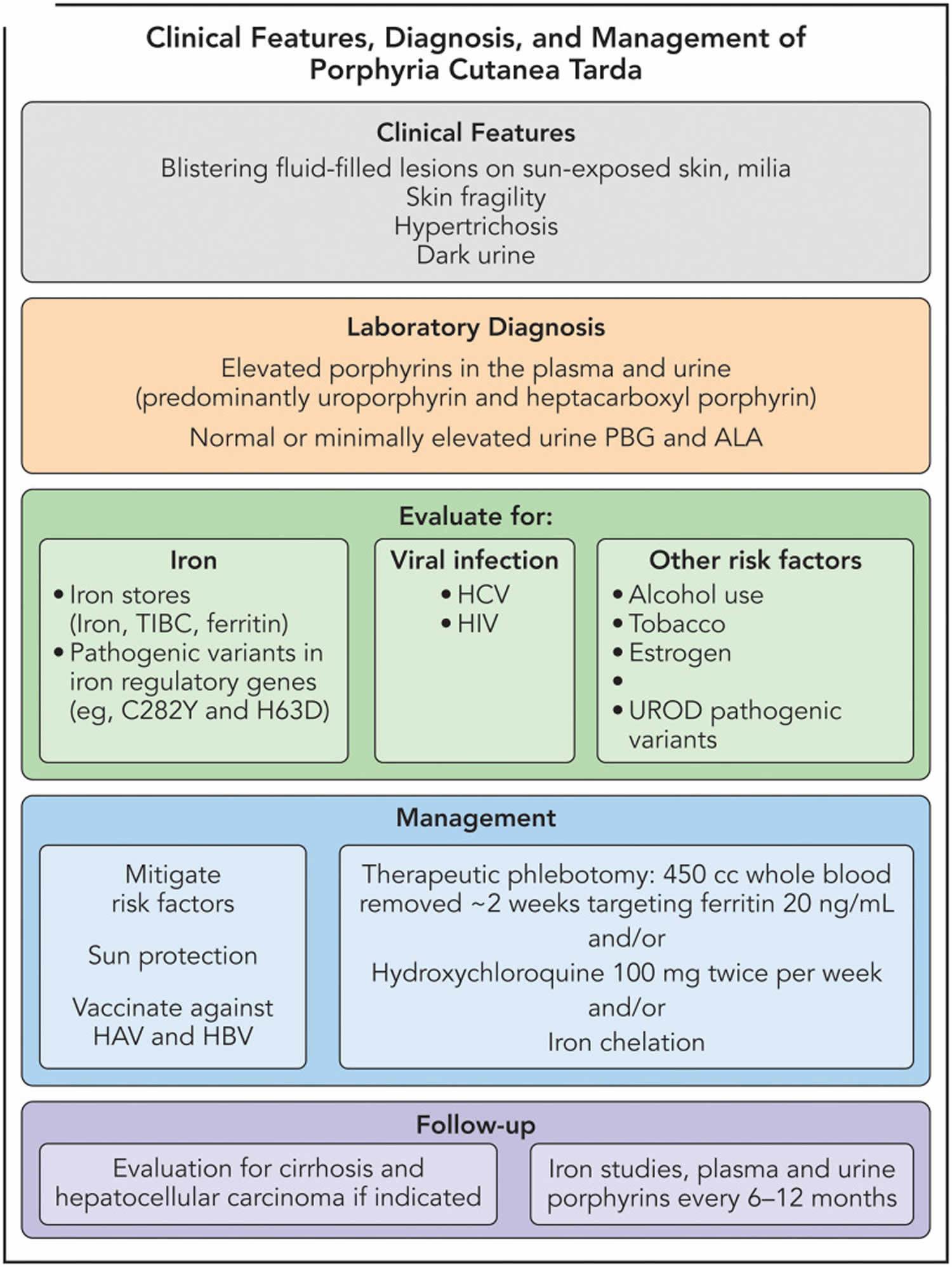
Abbreviations: HAV = hepatitis A virus; HBV = hepatitis B virus; PBG = porphobilinogen deaminase; TIBC = total iron-binding capacity.
[Source 12 ]Porphyria cutanea tarda causes
Porphyria cutanea tarda (PCT) is a multifactorial disorder, which means that several different factors such as genetic, infectious and environmental factors occurring in combination are necessary for the development of porphyria cutanea tarda 35. These factors are not necessarily the same for each individual. These factors contribute either directly or indirectly to decreased levels or ineffectiveness of an enzyme known as uroporphyrinogen decarboxylase (UROD) within your liver 36, 37, 38. When uroporphyrinogen decarboxylase (UROD) levels in your liver decrease to approximately 20% of normal levels, the symptoms of porphyria cutanea tarda may develop.
The uroporphyrinogen decarboxylase (UROD) enzyme is essential for breaking down (metabolizing) certain chemicals in the body known as porphyrins. Low levels of functional uroporphyrinogen decarboxylase (UROD) result in the abnormal accumulation of specific porphyrins in your body, especially within the blood, liver and skin. The symptoms of porphyria cutanea tarda occur because of this abnormal accumulation of porphyrins and related chemicals. For example when porphyrins accumulate in your skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin (photosensitivity) found in individuals with porphyria cutanea tarda. Your liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in your liver, they can cause toxic damage to your liver.
The exact, underlying mechanisms that cause porphyria cutanea tarda are complex and varied. It is determined that iron accumulation within the liver plays a central role in the development of the disorder in most individuals (sporadic or acquired porphyria cutanea tarda [PCT type 1]). Recently, researchers have discovered that a substance called uroporphomethene, which is an oxidized form of a specific porphyrin known as uroporphyrinogen, is an inhibitor that reduces the activity of the uroporphyrinogen decarboxylase (UROD) enzyme in the liver. The oxidation of uroporphyrinogen into uroporphomethene has been shown to be iron dependent, emphasizing the importance or elevated iron levels in the development of porphyria cutanea tarda.
The relationship between iron levels and porphyria cutanea tarda has long been established and porphyria cutanea tarda is classified as an iron-dependent disease. Clinical symptoms often correlate with abnormally elevated levels of iron in the liver (iron overloading). Iron overloading in the liver may only be mild or moderate. The exact relationship between iron accumulation and porphyria cutanea tarda is not fully understood, however, as there is no specific level of iron in the liver that correlates to disease in porphyria cutanea tarda (e.g. some individuals with symptomatic porphyria cutanea tarda have normal iron levels).
There is an increased prevalence of mutations in the HFE (homeostatic iron regulator) gene in individuals with porphyria cutanea tarda 26. The HFE gene provides instructions for producing a protein called HFE protein that is located on the surface of cells, primarily liver and intestinal cells 39. The HFE protein is also found on some immune system cells 39. The HFE protein interacts with other proteins on the cell surface to detect the amount of iron in the body. When the HFE protein is attached (bound) to a protein called transferrin receptor 1, the receptor cannot bind to a protein called transferrin. When transferrin receptor 1 is bound to transferrin, iron enters liver cells. So, it is likely that the HFE protein regulates iron levels in liver cells by preventing transferrin from binding to transferrin receptor 1. The HFE protein regulates the production of a protein called hepcidin 39. Hepcidin is produced by the liver, and it determines how much iron is absorbed from your diet and released from storage sites in your body 39. When the HFE protein is not bound to transferrin receptor 1, it binds to a group of other proteins that includes hepcidin. The formation of this protein complex (HFE protein+Hepcidin) triggers the production of hepcidin. So when the HFE protein is bound to transferrin receptor 1, hepcidin production is turned off and when the HFE protein is not bound to transferrin receptor 1, hepcidin production is turned on. When the proteins involved in iron sensing and absorption are functioning properly, iron absorption is tightly regulated. On average, the body absorbs about 10 percent of the iron obtained from the diet.
Mutations in the HFE gene can cause hemochromatosis, a genetic disorder causing the body to absorb and store excessive amounts of iron especially the liver, leading to potential organ damage 40. It’s also known as iron overload, and if left untreated, the excess iron can accumulate in vital organs like the liver, heart, and pancreas. Hemochromatosis occurs when a person inherited two mutated HFE genes (one from each parent). Hemochromatosis is associated with low levels of hepcidin, a specialized protein that is the primary regulator of iron absorption in the body, including regulating the uptake of iron by the gastrointestinal tract and liver.
Additional risk factors that have been associated with porphyria cutanea tarda include alcohol, certain infections such as hepatitis C or HIV, and drugs such as estrogens. Some studies have indicated that smoking is a risk factor for porphyria cutanea tarda in susceptible individuals. Less often, certain chemical exposures (e.g. hexachlorobenzene), kidney dialysis, and lupus appear to be connected to the development of porphyria cutanea tarda. It is believed that these susceptibility factors reduce hepcidin in the body and consequently lead to iron accumulation in the liver. However, the exact relationship among most susceptibility factors with the development of symptoms in porphyria cutanea tarda is not fully understood. For example, alcohol clearly contributes to the development of the disorder in some cases, but porphyria cutanea tarda is not common in alcoholics. Most individuals with porphyria cutanea tarda have three or more susceptibility factors present.
In some cases, individuals develop porphyria cutanea tarda without a known susceptibility factor, suggesting that additional, as yet unidentified risk factors exist.
The underlying cause of uroporphyrinogen decarboxylase deficiency in the acquired form of porphyria cutanea tarda is unknown. Affected individuals have approximately 50% residual uroporphyrinogen decarboxylase activity and do not develop symptoms unless additional factors are present. The most common factors associated with acquired porphyria cutanea tarda are hemochromatosis or chronic hepatitis C infection. In individuals with acquired porphyria cutanea tarda, uroporphyrinogen decarboxylase levels are only deficient in the liver.
In the familial form of porphyria cutanea tarda (familial porphyria cutanea tarda [PCT type 2]), individuals have a mutation in the uroporphyrinogen decarboxylase (UROD) gene. This mutation is inherited as an autosomal dominant trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new (de novo) mutation in the affected individual with no family history. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy regardless of the sex of the resulting child.
The uroporphyrinogen decarboxylase (UROD) gene creates (encodes) the uroporphyrinogen decarboxylase enzyme, which is the fifth enzyme in the heme synthesis pathway. A mutation in one of these genes leads to abnormally low levels of this enzyme in all tissues of the body (not just the liver). However, one mutation alone is insufficient to cause familial porphyria cutanea tarda (F-PCT) as residual uroporphyrinogen decarboxylase enzyme levels remain above 20% of normal. In fact, most individuals with a mutation in the uroporphyrinogen decarboxylase gene do not develop the disorder. Additional factors must be present for the disorder to develop.
Risk Factors for developing porphyria cutanea tarda
Mild to moderate iron overload alongside amplified serum ferritin levels and hepatic siderosis reportedly occurs in 90% of porphyria cutanea tarda cases 41, 42, 43. Iron overload can also be caused by mutations in the hemochromatosis gene (HFE gene) 44, 43. Hereditary hemochromatosis is a inherited disease causes the body to absorb too much iron from the diet triggering iron accumulation in organs such as liver 44, 45. The excess iron is stored in the body’s tissues and organs, particularly the skin, heart, liver, pancreas, and joints. Because humans cannot increase the excretion of iron, excess iron can overload and eventually damage tissues and organs. For this reason, hereditary hemochromatosis is also called an iron overload disorder. Early symptoms of hereditary hemochromatosis may include extreme tiredness (fatigue), joint pain, abdominal pain, weight loss, and loss of sex drive. As the condition worsens, affected individuals may develop arthritis, liver disease (cirrhosis) or liver cancer, diabetes, heart abnormalities, or skin discoloration. The appearance and severity of symptoms can be affected by environmental and lifestyle factors such as the amount of iron in the diet, alcohol use, and infections.
Mutations in several genes can cause hereditary hemochromatosis. Type 1 hemochromatosis results from mutations in the HFE gene, and type 2 hemochromatosis results from mutations in either the HJV or HAMP gene 44, 45. Mutations in the TFR2 gene cause type 3 hemochromatosis, and mutations in the SLC40A1 gene cause type 4 hemochromatosis 44, 45.
In type 1 hereditary hemochromatosis you inherit one HFE gene from each of your parents. The HFE gene has two common mutations, C282Y and H63D. Genetic testing can reveal whether you have these changes in your HFE gene. If you inherit two altered genes, you may develop hemochromatosis. You also can pass the altered gene on to your children. But not everyone who inherits two genes develops problems linked to the iron overload of hemochromatosis. If you inherit one altered gene, you’re unlikely to develop hemochromatosis. However, you are considered a carrier and can pass the altered gene on to your children. But your children wouldn’t develop hemochromatosis unless they also inherited another altered gene from the other parent.
Patients with porphyria cutanea tarda express more mutations in the HFE gene than the general population 41, 43. Data from several large studies indicate that the HFE gene mutation is present in almost 73% of porphyria cutanea tarda cases 41. Another study found 64.9% of porphyria cutanea tarda patients carried at least one HFE mutated allele 46. Other factors that can increase iron levels include hepatitis C virus infection, alcohol, and increased absorption of iron 41.
Heavy alcohol use (>40g/day) is recorded in almost 90% of porphyria cutanea tarda cases 41, and is more prevalent in males 46. Alcohol consumption exacerbates porphyria cutanea tarda by inhibiting the activity of delta-aminolevulinic acid dehydratase (ALAD), uroporphyrinogen decarboxylase (UROD), coproporphyrinogen-III oxidase (CPOX) and ferrochelatase (FECH), while enhancing the activity of aminolevulinate synthase (ALAS) and hydroxymethylbilane synthase (HMBS) thereby promoting accumulation of porphyrin 34. While the correlation between the effects of alcohol on aminolevulinate synthase (ALAS) and clinical expression of porphyria cutanea tarda is yet to be elucidated, chronic alcoholics are known to suffer from suppression of erythropoiesis and increased dietary iron absorption 29, 47. Alcohol is also thought to contribute to increased iron absorption, alcohol induced oxidative stress, and downregulation of hepcidin 41.
Smoking can induce earlier onset of Type 1 porphyria cutanea tarda (sporadic porphyria cutanea tarda) and is therefore a risk factor 48. Mechanisms of smoking mediating the development of porphyria cutanea tarda remain unclear, but increased oxidative stress and induction of hepatic cytochrome P450 enzymes are thought to contribute to disease pathology 41.
Infections with Hepatitis C Virus (HCV), and comorbid Hepatitis C Virus (HCV) and Human Immunodeficiency Virus (HIV) infections are associated with development of porphyria cutanea tarda 37. Hepatitis C Virus (HCV) is the most common porphyria cutanea tarda-related viral infection, and although associated with both subtypes, it is observed more frequently in Type 1 porphyria cutanea tarda (sporadic porphyria cutanea tarda) and Type 2 porphyria cutanea tarda (familial porphyria cutanea tarda) 49, 29, 50. A large study of 152 patients with porphyria cutanea tarda indicated that Hepatitis C Virus (HCV) infection is the most prevalent risk factor, especially in men 46. Although mechanisms are unclear, there is some indication that hepatitis C virus (HCV)-induced reactive oxygen species can trigger disease manifestations by fostering reduced hepcidin levels and promoting hepatic iron accumulation 51. Chronic hepatitis C virus (HCV) infection also lessens glutathione in hepatocytes, decreasing their ability to reduce oxidized porphyrins and causing their accumulation 29. It is important to note that hepatitis C virus (HCV)-infected persons develop porphyria cutanea tarda at an earlier age than those without the virus. HFE gene mutations also cause iron overload that further promote hepatocellular injury and fibrosis in patients with hepatitis C virus (HCV) 29.
Estrogens have been identified as precipitating factors for women with type 2 porphyria cutanea tarda (familial porphyria cutanea tarda [F-PCT]) 46, 37. Reports indicate use of oral contraceptives, hormone replacement therapy, and use of tamoxifen for breast cancer to be associated with porphyria cutanea tarda 41, 52, 53. Diethylstilbestrol, a synthetic nonsteroidal estrogen, also induces hepatic aminolevulinate synthase (ALAS), though there is currently no clear understanding of the accompanying increased porphyrin excretion in porphyria cutanea tarda patients 29. Estrogen as a treatment for prostate cancer has also been identified as a risk factor in men 54. Administration of estrogens via transdermal route is safe and recommended for at-risk women previously treated for porphyria cutanea tarda 53.
Hepatic siderosis (hepatic iron overload or the abnormal accumulation of iron in the liver caused by various factors, including hereditary hemochromatosis and secondary causes like chronic liver disease or repeated blood transfusions), systemic lupus erythematosus (SLE), end-stage renal disease on hemodialysis, diabetes mellitus, and hematologic malignancies are all associated with the development of porphyria cutanea tarda 55, 53, 56, 57, 58, 59, 41, 29.
Exposure to toxins such as polychlorinated biphenyls, hexachlorobenzene, and other polyhalogenated hydrocarbons that significantly induce cytochrome P450 enzymes are also associated with the development of porphyria cutanea tarda 28.
Porphyria cutanea tarda pathophysiology
Porphyria cutanea tarda results from the inhibition of liver uroporphyrinogen decarboxylase (UROD) enzyme, a cytoplasmic housekeeping enzyme that converts uroporphyrinogen to coproporphyrinogen 12. The uroporphyrinogen decarboxylase (UROD) enzyme is encoded by the UROD gene, located on chromosome 1 with 10 exons spanning over 3 kb 60, 61. The uroporphyrinogen decarboxylase (UROD) enzyme carries out a complex reaction, sequentially decarboxylating the 4 acetyl groups of uroporphyrinogen (an octacarboxyl porphyrin) to hepta-, hexa-, penta-, and finally coproporphyrinogen (a tetracarboxyl porphyrin) 41, 62. Both uroporphyrinogen I and III isomers are decarboxylated by uroporphyrinogen decarboxylase (UROD), but uroporphyrinogen III is preferred because coproporphyrinogen oxidase is specific for coproporphyrinogen III, and the III isomers are intermediates in heme synthesis 63, 64, 65, 65.
The liver uroporphyrinogen decarboxylase (UROD) protein level remains at its genetically determined level in all types of porphyria cutanea tarda, but liver enzyme activity is reduced to less than about 20% of normal, suggesting the presence of an enzyme inhibitor 12. Phillips and colleagues 20 identified this inhibitor as a uroporphomethene, probably derived from the partial oxidization of uroporphyrinogen. At least in mice models, cytochrome P450 enzyme activity is involved in the generation of uroporphomethene inhibitor. Uroporphomethene differs from uroporphyrinogen by a single oxidized bridge carbon, and although it is able to bind strongly to the active site of UROD, it is unable to serve as a substrate 20 However, other researchers have questioned whether uroporphomethene is in fact a true inhibitor of UROD based on its fragmentation pattern on mass spectrometry 66.
When hepatic UROD activity is reduced to less than 20% of normal activity, uroporphyrinogen and the porphyrinogens that are intermediates in its 4-step decarboxylation accumulate in the liver and are auto-oxidized to their corresponding porphyrins 67. After considerable accumulation in the liver, these porphyrins (uroporphyrinogen and porphyrinogens) appear in plasma and bile and are excreted in the urine and stool 63. These porphyrins are activated by light exposure (especially at wavelengths near 400 nm) and generate reactive oxygen species that damage sun-exposed skin 67. Furthermore, in UROD-deficient mice, the upregulation of delta-aminolevulinic acid synthase 1 (ALAS-1) by drugs that induce hepatic P450 enzymes and the supplementation of δ-aminolevulinic acid (ALA) in the drinking water have been shown to induce a porphyria cutanea tarda phenotype 68.
Porphyria cutanea tarda symptoms
The symptoms of porphyria cutanea tarda (PCT) can vary greatly from one individual to another. The symptoms of porphyria cutanea tarda are confined mostly to your skin. Excess porphyrin in your skin results in photosensitivity (a condition in which the skin becomes very sensitive to sunlight or other forms of ultraviolet light and may burn easily). Individuals with porphyria cutanea tarda (PCT) develop blisters on sun-exposed areas of their skin (photosensitivity), such as the back of the hands and the forearms. Other sun-exposed sites such as the face, scalp, neck, and arms may also be affected. The skin in these areas may blister or peel after minor trauma. Eventually, scarring may develop and affected skin may darken (hyperpigmentation) or fade (hypopigmentation) in color. Abnormal, excessive hair growth (hypertrichosis), especially on the face may also occur. The hair may be very fine or coarse and can differ in color. In some patients, their hair may grow, thicken and darken. Small bumps with a distinct white head (milia) may also develop, especially on the backs of the hands. In some cases, the skin in affected areas may thickened and harden, resembling a condition known as sclerosis, this is sometimes known as pseudosclerosis. Pseudosclerosis in individuals with porphyria cutanea tarda appears as scattered, waxy, harden patches or plaques of skin. Characteristically, the urine is darker than usual, with a reddish or tea-colored hue.
Neurological and abdominal symptoms are not characteristic of porphyria cutanea tarda.
Liver abnormalities may develop in some affected individuals including the accumulation of iron in the liver (hepatic siderosis), the accumulation of fat in the liver (steatosis), inflammation of certain parts of the liver (portal triaditis), and thickening and scarring around the portal vein (periportal fibrosis). Affected individuals may be at a greater risk than the general population of developing scarring of the liver (cirrhosis) or liver cancer known as hepatocellular carcinoma (HCC). Advanced liver disease is uncommon, except in older individuals with recurrent disease. In some cases, liver disease is due to an associated condition such as hepatitis B or hepatitis C infection.
Porphyria cutanea tarda, Hepatitis C Virus and HIV
Because porphyria cutanea tarda is frequently associated with hepatitis C virus infection, it is worth noting the issues involved in treating a patient with both porphyria cutanea tarda and hepatitis C virus infection.
Infection with hepatitis C virus is much more common than porphyria cutanea tarda, and most people with hepatitis C virus do not have porphyria cutanea tarda. However, at least in some locations, as many as 80 percent of individuals with porphyria cutanea tarda are infected with hepatitis C virus. Therefore, hepatitis C virus needs to be added to the list of factors that can activate porphyria cutanea tarda alongside alcohol, iron and estrogens. Other hepatitis viruses are seldom implicated in porphyria cutanea tarda, and it is not known how hepatitis C virus activates porphyria cutanea tarda.
There are several different viruses that cause hepatitis. A blood test for hepatitis C virus infection has not been available for very long. Hepatitis C virus is most readily transmitted from one person to another by blood products. Although most people who are infected with hepatitis C virus have a history of exposure to blood or needles contaminated with blood, in some cases it is not known how the infection was acquired. Hepatitis C virus (unlike the hepatitis B Virus and HIV) is seldom transmitted by sexual contact. It is also not readily transmitted by casual contact with other people. Therefore, people infected with hepatitis C virus are not hazardous unless they somehow expose others to their blood.
It is recommended that patients with porphyria cutanea tarda be tested for hepatitis C virus infection. This is done by a blood test that detects antibodies to the virus. If hepatitis C virus infection is found, it may not change the treatment of porphyria cutanea tarda (by phlebotomy or low-dose chloroquine). Treatment for porphyria cutanea tarda is highly successful even in patients with hepatitis C virus. Therefore, it is reasonable to treat the porphyria cutanea tarda first and then look into treatment for hepatitis C virus later.
There are reasons not to treat the hepatitis C virus infection before treating the porphyria cutanea tarda. Hepatitis C virus treatment with alpha-interferon and ribavirin is available but is often not effective. Also, liver damage progresses slowly if at all in many people with hepatitis C virus. However, once the porphyria cutanea tarda is in remission it is important to assess the amount of liver damage the virus has already caused and to have follow-up visits to a doctor to monitor the liver. In some cases it may be important to treat hepatitis C virus infection to try and prevent progressive liver damage.
Porphyria cutanea tarda complications
Although the signs and symptoms of porphyria cutanea tarda are limited to the skin, patients are also at a high risk of liver complications. Liver biopsies usually reveal fatty changes along with porphyrin deposits and sometimes lobular necrosis. Porphyria cutanea tarda independently increases the risk of liver cirrhosis and hepatocellular carcinoma, which may be accentuated by co-existing hepatitis C virus (HCV) infection, alcoholic hepatitis, or iron overload 69.
Porphyria cutanea tarda diagnosis
Porphyria cutanea tarda (PCT) may be clinically suspected but should always be confirmed by laboratory tests. The preferred screening test for porphyria cutanea tarda is a measurement of porphyrins in plasma and urin. This can differentiate porphyria cutanea tarda (PCT) from Variegate Porphyria (VP). Urine and feces need to be sent to analyse the porphyrin levels, which will be elevated. The specimens need to be protected from light with aluminium foil to ensure testing is accurate. Examination of the urine with a Wood’s lamp (Wood’s lamp emits long-wave ultraviolet (UV) light) may reveal coral pink fluorescence due to excessive porphyrins.
Porphyria cutanea tarda (PCT) is diagnosed biochemically by high levels of porphyrins in the plasma and urine, with a predominance of uroporphyrinogen and hepta- and hexa- and pentacarboxyl porphyrins. This pattern of porphyrin elevation is characteristic but not completely specific since uroporphyrin elevation occurs in other porphyrias, and a PCT-like pattern occurs in some patients with variegate porphyria (VP) 70. Therefore, analysis of porphyrins in the red blood cells (erythrocytes) and feces should be considered. Urine measurements using random urine samples with normalization to creatinine is recommended. Urine porphobilinogen is normal in porphyria cutanea tarda (PCT), and delta-aminolevulinic acid (ALA) is normal or only mildly increased 41. The patterns of porphyrins in urine (predominately uroporphyrin and 7-carboxylate porphyrin) and feces (predominately isocoproporphyrin) help to confirm the diagnosis. Plasma fluorescence scanning is useful for rapid differentiation of variegate porphyria (VP), which has a diagnostic peak at approximately 626 nm. Fecal total porphyrins may be normal or elevated in porphyria cutanea tarda (PCT), and an elevation of fecal isocoproporphyrins is specific for UROD inhibition 70, 71.
Familial porphyria cutanea tarda (F-PCT), an inherited deficiency of uroporphyrinogen decarboxylase (UROD) enzyme, can be diagnosed by the presence of a reduced amount of the uroporphyrinogen decarboxylase (UROD) enzyme in red blood cells (erythrocytes) and is present in about 20% of patients with porphyria cutanea tarda 63. Molecular genetic testing is available for familial porphyria cutanea tarda if the diagnosis has been confirmed in the patient or a family member by urinary porphyrin analysis and/or enzyme assay of uroporphyrinogen decarboxylase activity 72, 73.
A skin biopsy can be helpful to distinguish porphyria cutanea tarda (PCT) from other skin blistering conditions. The skin changes are identical to Variegate Porphyria (VP) and Hereditary Coproporphyria (HCP).
Tests to determine the cause of the porphyria may include:
- Blood count, liver function, and kidney function tests
- Iron studies (ferritin level)
- Hepatitis B, C, and human immunodeficiency virus (HIV) serology
- Transferrin saturation and genotyping for HFE gene mutations for hereditary hemochromatosis 44
- Tests for cutaneous lupus erythematosus and diabetes
- Urodecarboxylase (UROD) enzyme levels and genetic tests.
Porphyria cutanea tarda treatment
Porphyria cutanea tarda is the most treatable of the porphyrias. The treatment of porphyria cutanea tarda is directed toward the underlying liver problem and the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, general internists, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan your treatment.
The first step in the management of porphyria cutanea tarda is the avoidance of all risk factors such as strictly avoiding alcohol, smoking, and estrogen therapy, along with limiting any excess intake of iron. Since porphyria cutanea tarda (PCT) is a photosensitive skin condition, sunlight avoidance is the key until the porphyrin levels have normalized. The wavelengths inducing porphyrins are in the range of 400-410 nm, and only titanium dioxide or zinc oxide containing sunscreen is effective 27. Protective clothing is also helpful in protecting the skin from harmful sunlight rays. Any affected skin areas should be kept clean to prevent the development of skin infections, and associated pain can be managed with oral analgesics.
Presently, there are no effective treatments that restore UROD enzyme levels in individuals with familial porphyria cutanea tarda (F-PCT) 28. However, treatment seems to be equally effective in familial porphyria cutanea tarda (F-PCT) and non-familial porphyria cutanea tarda. Factors that tend to activate the disease should be removed and may result in the resolution of porphyria cutanea tarda. Treatment may include reducing alcohol consumption, stopping estrogen or hormone treatment, avoiding excessive iron intake, or antiviral treatment for underlying hepatitis C.
Reduction of liver iron content is the general recommendation and the most widely recommended treatment is a schedule of repeated phlebotomies (removal of blood), with the aim of reducing iron in the liver 28, 23, 29, 30, 31. This actually reduces iron stores throughout the body. Usually, removal of only 5 to 6 pints of blood (one pint [approximately 450 ml of blood] every one to two weeks) is sufficient, which indicates that iron stores are not excessively increased in most porphyria cutanea tarda patients. The best guides to response are measurements of serum ferritin and plasma porphyrins. Phlebotomies are stopped when the ferritin falls to ~20 ng/ml 32, 33. Normal ferritin levels vary by gender and age, but generally, for adult males, ferritin level is between 30-300 ng/mL, and for adult females, it’s between 13-150 ng/mL.
Iron chelation therapy i.e., deferasirox or deferoxamine may be considered when phlebotomy is contraindicated, and low iron diet may be beneficial if the latter fails 28.
If phlebotomy cannot be done, as in elderly patients or those who are anemic, antimalarial tablets such as low doses of either chloroquine (125mg twice weekly) or hydroxychloroquine (100mg twice weekly) to allow the porphyrins to be excreted more easily. Usual dosages of these drugs should not be used because they can cause transient but sometimes severe liver damage and worsening of photosensitivity in porphyria cutanea tarda patients.
Furthermore, use of antiviral therapy may benefit patients with chronic hepatitis C infections and reduce risk of progressing to liver cancer 29.
After treatment for porphyria cutanea tarda, periodic measurement of plasma porphyrins may be advised, especially if a contributing factor such as estrogen exposure is resumed. If a recurrence does occur, it can be detected early and treated promptly. The treatment of porphyria cutanea tarda is almost always successful, and the prognosis is usually excellent.
Phlebotomy
Any condition leading to iron overload in the patient is a clear indication for phlebotomy, and in porphyria cutanea tarda cases, phlebotomy is preferred over hydroxychloroquine. Different protocols have been tried, such as removing one unit or 450 ml of blood every two weeks. Strict serial monitoring of ferritin levels is done, and a downward trend in serum ferritin level is the goal of phlebotomy (till a ferritin level of less than 20 ng/ml is seen) 32, 33. Alternatively, 300ml of blood removed weekly is another treatment strategy that can be used. Care should be taken not to induce anemia or hemoglobin less than 10 gm/dL. Contraindications to phlebotomy include patients with pulmonary or coronary artery disease.
The skin manifestations resolve within four months, but the porphyrin levels can take up to 12 months to normalize 23. Serum ferritin levels may be used as an indicator to monitor for relapse of porphyria cutanea tarda since the levels change before porphyrin levels 74.
Hydroxychloroquine or chloroquine
Iron chelation therapy
Iron chelation was considered to be an alternative in people with iron overload-induced porphyria cutanea tarda, but after comparative studies with phlebotomy and hydroxychloroquine, it was not found to be as efficient 23. However, deferoxamine and deferasirox can be used in patients with contraindications to both phlebotomy and hydroxychloroquine 23. The disadvantages of iron chelation therapy other than being expensive due to the use of a subcutaneous pump were the failure to normalize the porphyrin level even after 12 months of treatment 23.
Porphyria cutanea tarda prognosis
Porphyria cutanea tarda is the most treatable of the porphyrias. Once clear, porphyria cutanea tarda is unlikely to recur unless the underlying risk factors (susceptibility factors) have not been addressed. Relapses of up to 35% have been recorded over a follow-up period of up to 11 years 76. People with porphyria cutanea tarda with elevated iron levels may need periodic phlebotomy (removal of blood). Patients who continue to be exposed to risk factors (susceptibility factors), such as excessive alcohol consumption and smoking, are also more likely to relapse 23. Annual monitoring of urine and plasma uroporphyrin levels is recommended to detect biochemical relapses before the clinical manifestations of the disease appear 49. If porphyria cutanea tarda is ongoing, there can be an increased risk of developing hepatocellular carcinoma (HCC) or liver cancer, especially in populations of older men with long-standing active disease, heavy alcohol intake, and cirrhosis 77. Most of the studies predate recognition of hepatitis C prevalence in populations with porphyria cutanea tarda or hepatocellular carcinoma (HCC); many reported liver cancers may have been, at least in part, as complication of chronic hepatitis C infection 78.
Hepatoerythropoietic Porphyria
Hepatoerythropoietic porphyria (HEP) also called UROD-related hepatoerythropoietic porphyria is an extremely rare inherited disorder of the heme-biosynthetic pathway known as cutaneous porphyrias caused by mutations on both copies of a person’s UROD gene that encodes uroporphyrinogen decarboxylase (UROD) enzyme that is crucial in the fifth step of the heme biosynthesis pathway, which means that hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait 79, 80, 34, 81, 82, 83, 84, 85, 86, 87, 88, 89, 90, 91, 92, 93. In hepatoerythropoietic porphyria (HEP), the uroporphyrinogen decarboxylase (UROD) enzyme activity is usually less than 10% its normal levels 94. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in the body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. When porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with hepatoerythropoietic porphyria (HEP). The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
Most affected individuals with hepatoerythropoietic porphyria (HEP) have a profound deficiency of uroporphyrinogen decarboxylase (UROD) enzyme and onset of hepatoerythropoietic porphyria (HEP) is usually during infancy or early childhood. However, some individuals may have a mild form that can go undiagnosed until adulthood. The childhood form of hepatoerythropoietic porphyria (HEP) is often associated skin photosensitivity with painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected areas of skin can scar often with mutilation and loss of facial features and fingers and become discolored 92. Bacteria may infect the damaged skin and contribute to mutilation and scarring. The signs and symptoms of childhood form of hepatoerythropoietic porphyria (HEP) resemble congenital erythropoietic porphyria (CEP), with symptoms of skin blistering on sun-exposed skin that usually begin in infancy. Abnormal, excessive hair (hypertrichosis), red-brown discoloration of teeth (erythrodontia), and reddish-colored urine. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D deficiency. Red blood cells have a shortened life-span with mild or severe hemolytic anemia. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with abnormal enlargement of the liver and/or spleen (hepatosplenomegaly). Mild cases of hepatoerythropoietic porphyria (HEP) may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), the most common form of porphyria in humans where the porphyria cutanea tarda (PCT) that may be acquired in 75% to 80% of cases (also known as porphyria cutanea tarda type 1 or sporadic porphyria cutanea tarda) or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance or porphyria cutanea tarda type 2 or familial porphyria cutanea tarda). Hepatoerythropoietic Porphyria (HEP) is the autosomal recessive form of familial porphyria cutanea tarda (F-PCT). Skin photosensitivity is generally much more severe in hepatoerythropoietic porphyria (HEP) than in porphyria cutanea tarda (PCT).
Hepatoerythropoietic porphyria (HEP) is an extremely rare disorder that affects males and females in equal numbers. Approximately less than 100 cases have been reported in the medical literature 34, 95, 96, 97. The exact incidence or prevalence of hepatoerythropoietic porphyria (HEP) in the general population is unknown 97. The frequency of hepatoerythropoietic porphyria (HEP) can only be inferred based on that of familial porphyria cutanea tarda (F-PCT), which occurs in one in 20,000 individuals 79. Over a ten-year period from 2007 to 2017, a referral center or porphyria-specific diagnostic laboratory provided molecular diagnostic testing on 4 unrelated individuals with hepatoerythropoietic porphyria (HEP), identifying one novel variant 98. Two founder variants have been identified in Norway 99.
A diagnosis of hepatoerythropoietic porphyria (HEP) is based upon identification of characteristic symptoms, a detailed medical history, a thorough clinical evaluation and a variety of specialized tests. Hepatoerythropoietic porphyria (HEP) may be considered in infants and children with chronic, blistering photosensitivity.
Diagnosis of hepatoerythropoietic porphyria (HEP) can be made by demonstrating significant elevations of specific porphyrins in your urine and stool, as well as identification of a specific fluorescence emission peak in plasma. DNA genetic testing to identify the specific mutations in an individual’s UROD genes is the most specific and sensitive test to confirm the diagnosis of hepatoerythropoietic porphyria (HEP).
The treatment of hepatoerythropoietic porphyria (HEP) is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with hepatoerythropoietic porphyria (HEP). Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance and/or protection from sunlight will benefit individuals with hepatoerythropoietic porphyria (HEP) and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies (removal of blood) to lower the amount of porphyrins in the liver, which are used to treat individuals with porphyria cutanea tarda (PCT), are generally ineffective in individuals with hepatoerythropoietic porphyria (HEP) since elevated iron levels are not a feature of the disorder. Another treatment for porphyria cutanea tarda (PCT), the antimalarial drug hydroxychloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin (rEPO) a synthetic version of the naturally occurring hormone erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with hepatoerythropoietic porphyria (HEP) whose anemia was not associated with increased red cell destruction.
Figure 11. Hepatoerythropoietic porphyria (HEP)
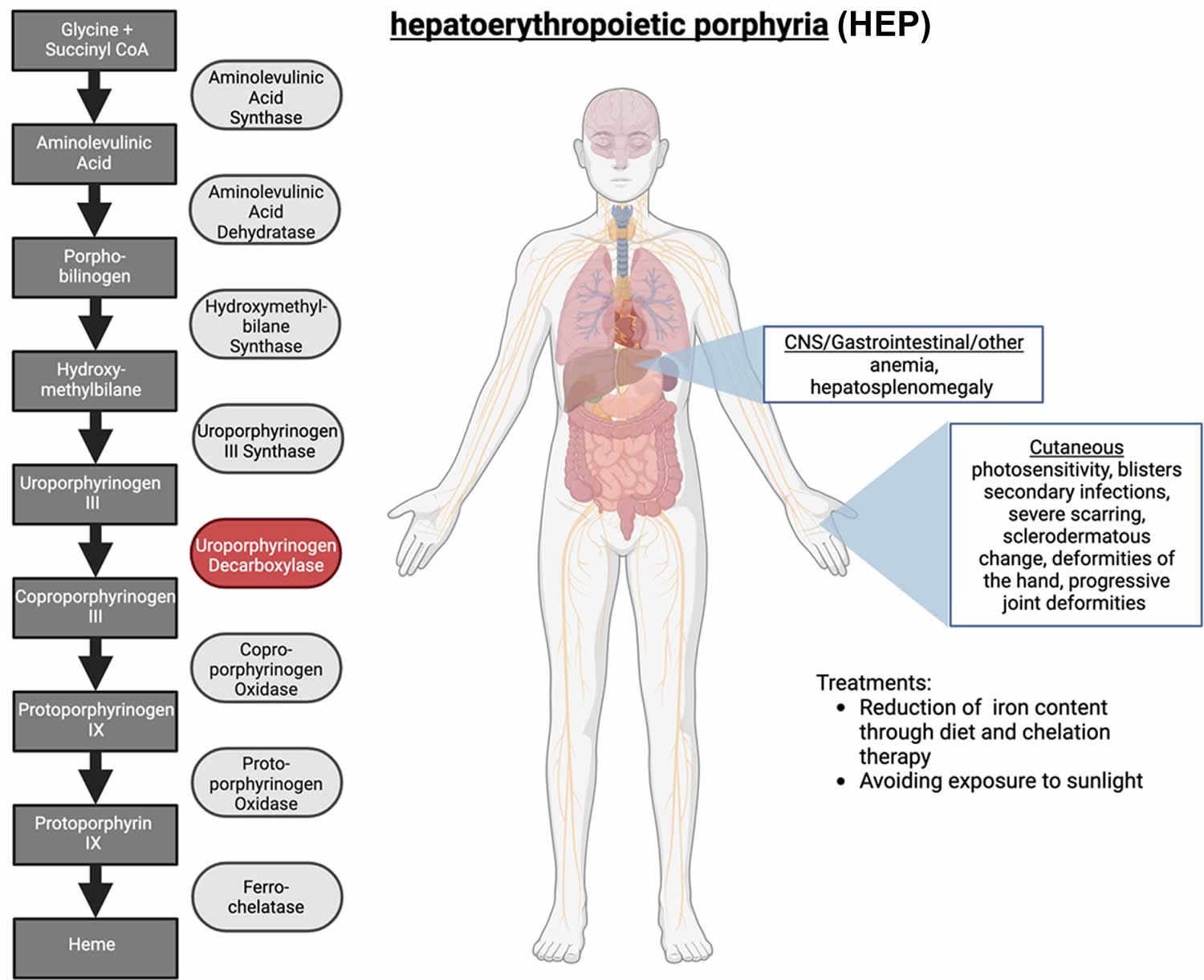
Figure 12. Hepatoerythropoietic porphyria (HEP)
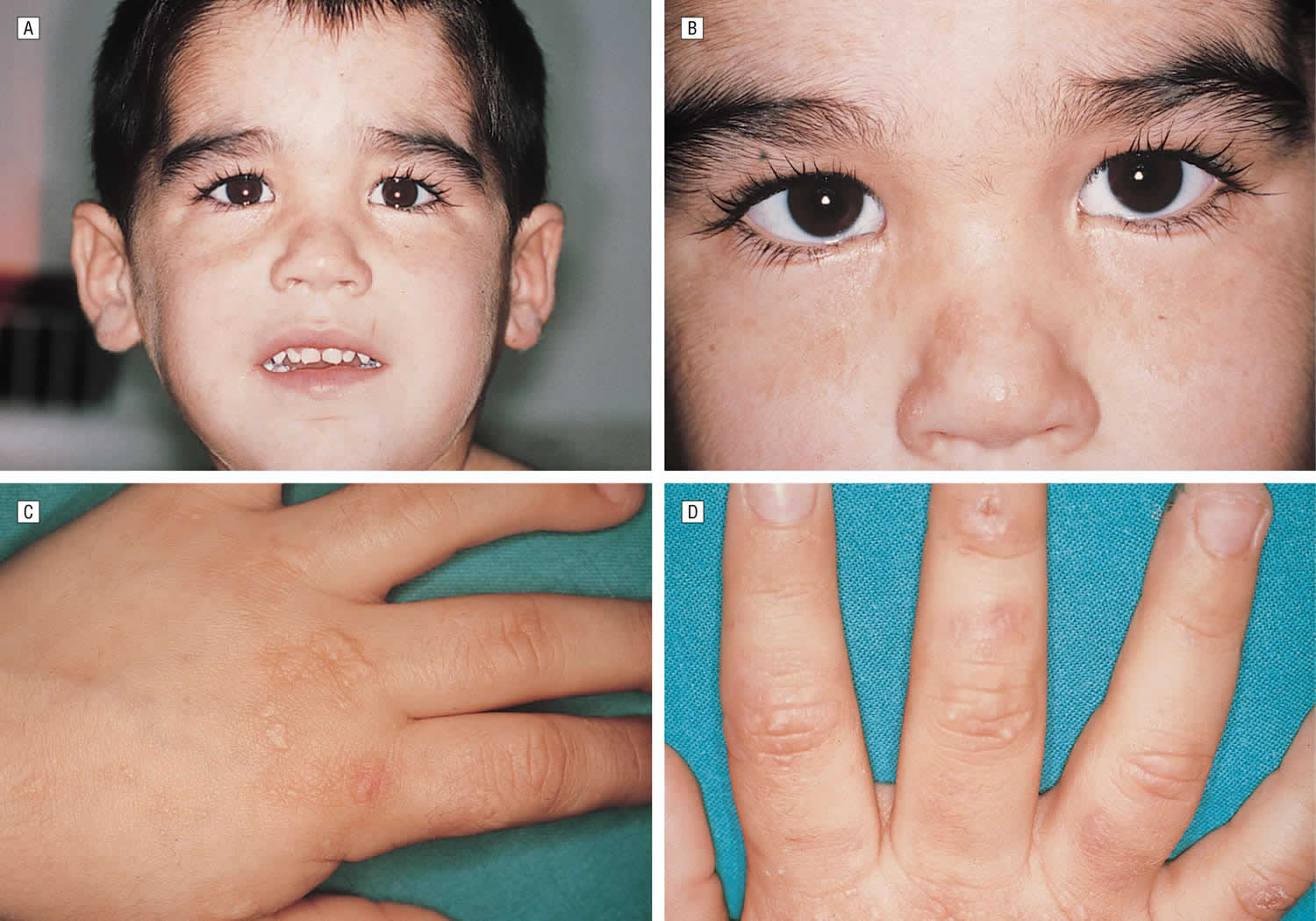
Footnotes: A 5-year-old boy, with no relevant personal or family history, had a syndrome of skin hyperfragility and photosensitivity since 2 years of age. His urine was dark. Skin lesions appeared as vesicles, blisters, and erosions on the face and the back of the hands. Lesions resolved with superficial scars and milia cysts. He presented with hypertrichosis on the face, limbs, and trunk. In the past 3 years, the patient has not presented with any active lesions, and only some superficial scars and mild hypertrichosis remained visible. (A) and (B), The patient, aged 5 years, with mild hypertrichosis of the forehead. (C) and (D) Superficial scars, after blisters and erosions, on the back of the hands. The young boy had a typical profile of porphyrin accumulation with an excess of urinary uroporphyrin and the presence of isocoproporphyrin in feces. Erythrocytic UROD catalytic activity was dramatically decreased. The diagnosis of hepatoerythropoietic porphyria (HEP) was therefore made and confirmed by UROD gene analysis. A point mutation in the third exon of the UROD gene was found at codon 46, a phenylalanine-to-leucine substitution (F46L). The boy was homozygous and the parents were heterozygous for the mutation.
[Source 86 ]Figure 13. Hepatoerythropoietic Porphyria dark urine

Hepatoerythropoietic porphyria cause
Hepatoerythropoietic porphyria (HEP) is caused by mutations on both copies of a person’s UROD gene that provides instructions for making an enzyme known as uroporphyrinogen decarboxylase (UROD) that is crucial in the fifth step of the heme biosynthesis pathway, which means that hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait 79, 82. Heme (haem) is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). The production of heme (haem) is a multi-step process that requires 8 different enzymes. Uroporphyrinogen decarboxylase (UROD) is responsible for the fifth step in heme biosynthesis process, in which carbon and oxygen atoms are removed from uroporphyrinogen III (the product of the fourth step) to form coproporphyrinogen III. In subsequent steps, three other enzymes produce and modify compounds that ultimately lead to heme.
Scientists have determined that the UROD gene is located on the short arm (p) of chromosome 1 (1p34.1) 97. Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 also known as autosomes and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 1p34.1” refers to band 34.1 on the short arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome. At least 30 different mutations of the UROD gene have been identified in patients with hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT), with 1 predominant missense mutation (glycine–to–glutamic acid substitution at codon 281) in Spanish patients with hepatoerythropoietic porphyria (HEP) 86.
In hepatoerythropoietic porphyria (HEP), the uroporphyrinogen decarboxylase (UROD) enzyme activity is usually less than 10% its normal levels 97. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in the body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. When porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with hepatoerythropoietic porphyria (HEP) 97. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
The rarity of hepatoerythropoietic porphyria (HEP) makes identification of additional risk factors difficult to assess 79. However, the existence of instances of late-onset disease suggest that risk factors may play a role in some hepatoerythropoietic porphyria (HEP) individuals 100. Excess iron may contribute to UROD inhibition by providing an oxidative environment that is apparently required for generating a UROD inhibitor 30. Hepatic hepcidin expression has been shown to regulate iron homeostasis and likely plays a role in development of porphyria cutanea tarda (PCT); however, the role that
hepcidin plays in porphyria cutanea tarda (PCT) development has not been clearly defined 55.
Alcohol and its metabolites may induce the enzymes ALAS1 and CYP2E1, generate reactive oxygen species (ROS) that contribute to oxidative damage, cause mitochondrial injury, deplete reduced glutathione and other antioxidant defenses, increase endotoxin production, and activate Kupffer cells leading to liver inflammation. In addition, alcohol has been found to impair iron-mediated expression of hepatic hepcidin and to decrease hepatic expression of hepcidin, which may help lead to increased iron in hepatocytes 101, 102.
Smoking may increase oxidative stress in hepatocytes and induce hepatic CYP1A2 which is important in the development of uroporphyria in rodent models. Hepatitis C is associated with excess fat, some iron accumulation, mitochondrial dysfunction, and oxidative stress in hepatocytes – all of which may contribute to the development of porphyria cutanea tarda (PCT). Dysregulation of hepcidin may contribute to iron accumulation in hepatitis C 103, 51.
Estrogens can generate reactive oxygen species (ROS) in some experimental systems; however, the mechanism by which they are a susceptibility factor has not been established. Estrogen mimetics (e.g., tamoxifen) have been shown to be associated with porphyria cutanea tarda (PCT) in several cases. The liver is the site of estrogen metabolism; first pass kinetics leads to a much higher hepatic concentration of estrogen and may also contribute to an increased oxidative environment in some individuals 53.
Hepatoerythropoietic porphyria inheritance pattern
Hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 14. Hepatoerythropoietic porphyria autosomal recessive inheritance pattern
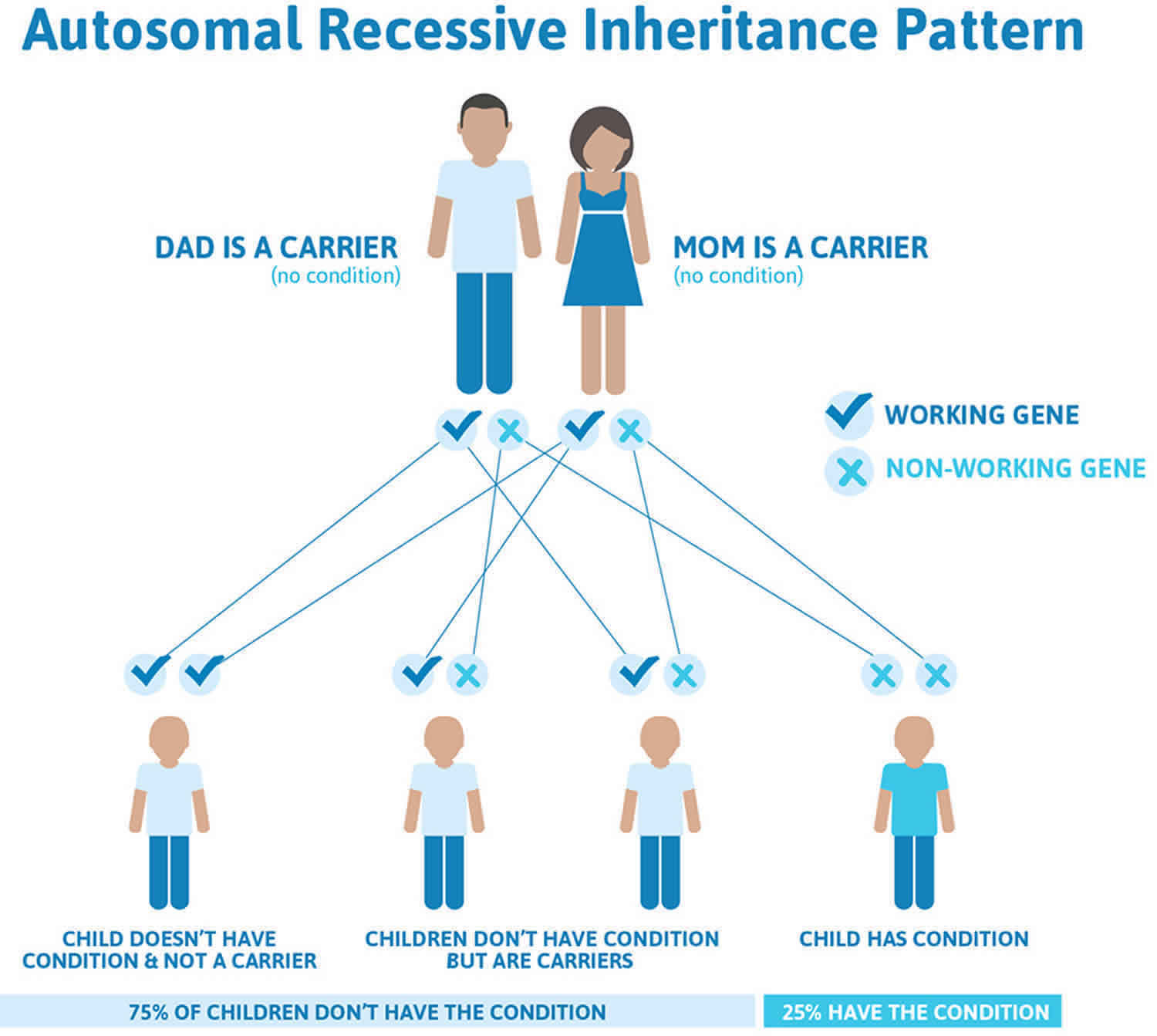
Hepatoerythropoietic porphyria pathophysiology
Reduced activity of the liver enzyme uroporphyrinogen decarboxylase (UROD) to 15%-20% of normal in all tissues leads to the accumulation of substrate, uroporphyrinogen, and the intermediate products of the reaction in all cells. Cells with a high demand for heme production include red blood cells (erythrocytes) and liver cells (hepatocytes); therefore, accumulation is more pronounced in these cell types predominantly in the liver.
The substrates uroporphyrinogen and intermediates accumulate in cells in the form of oxidized porphyrins mostly uroporphyrin and heptacarboxylporphyrin that are then transported into the plasma, where they are deposited in the skin and other tissues. In the skin, these porphyrins interact with blue light (the Soret band, ~410 nm) to produce skin damage.
Excess plasma porphyrins (i.e., uroporphyrin and heptacarboxylporphyrin) are excreted via the urine. Intermediates further along in the pathway (i.e., protoporphyrin and zinc protoporphyrin) are eliminated in the bile.
Hepatoerythropoietic porphyria sgns and symptoms
Hepatoerythropoietic porphyria (HEP) symptoms and severity can vary from one person to another. Onset is usually within the first two years of life, but mild cases that go undiagnosed until adulthood have been reported. Although hepatoerythropoietic porphyria (HEP) is associated with specific, characteristic symptoms, several factors, including the small number of identified cases, make it difficult to establish the full range of associated symptoms of hepatoerythropoietic porphyria (HEP).
Severe skin photosensitivity is usually the first sign. Affected infants may have extremely fragile skin that can peel or blister on minimal impact is common. Reddening of the skin is also common (erythema). Blistering skin lesions can develop on sun-exposed skin such as the hands and face. Photosensitivity can be severe and can cause scarring, erosion, and disfigurement. Bacterial infection of skin lesions can occur. Abnormal, excessive hair growth (hypertrichosis) may also occur on sun-exposed skin. Affected skin may darken or lose color (hyper- or hypopigmentation). Small bumps with a distinct white head (milia) may also develop. Some affected individuals have teeth that are reddish-brown colored (erythrodontia) as a result of the deposition of porphyrins in the enamel layer of the developing tooth 79.
Low levels of circulating red blood cells (anemia) may also occur. Anemia may be due to the premature destruction of red blood cells (hemolysis). Anemia associated with hepatoerythropoietic porphyria (HEP) may be mild or severe. Severe anemia may be associated with fatigue, pale skin, irregular heartbeat, chest pain, dizziness, and abnormally cold hands and feet. Some individuals may have an abnormally enlarged liver and/or spleen (hepatosplenomegaly).
Mild cases of hepatoerythropoietic porphyria (HEP) can go undiagnosed until adulthood. Overt photosensitivity may not be seen, and mild skin damage can be mistaken for other conditions during childhood.
Note: The clinical features of hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT) are indistinguishable. However, in striking contrast to hepatoerythropoietic porphyria (HEP), familial porphyria cutanea tarda (F-PCT) rarely if ever manifests in infancy or childhood. Rather, as is the case for porphyria cutanea tarda type 1 (nonfamilial, acquired), familial porphyria cutanea tarda (F-PCT) is a disease of middle-aged persons, usually with several risk factors for development of overt disease.
Hepatoerythropoietic porphyria diagnosis
A diagnosis of hepatoerythropoietic porphyria (HEP) is based upon identification of characteristic symptoms, a detailed medical history, a thorough clinical evaluation and a variety of specialized tests.
Hepatoerythropoietic porphyria (HEP) should be suspected in infants or children with the following clinical findings, suggestive laboratory findings, and family history.
Hepatoerythropoietic porphyria (HEP) may be considered in infants and children with these clinical features:
- Blistering skin lesions/vesicles/bullae
- Abnormal, excessive hair (hypertrichosis)
- Scarring
- Passage of red or dark urine
Note: The features of hepatoerythropoietic porphyria (HEP) generally resemble those of congenital erythropoietic porphyria (CEP).
Diagnosis of hepatoerythropoietic porphyria (HEP) can be made by demonstrating significant elevations of specific porphyrins in your urine and stool, as well as identification of a specific fluorescence emission peak in plasma. DNA genetic testing to identify the specific mutations in an individual’s UROD genes is the most specific and sensitive test to confirm the diagnosis of hepatoerythropoietic porphyria (HEP).
Clinical Testing and Workup
Screening tests can help diagnose hepatoerythropoietic porphyria (HEP) by measuring the levels of certain porphyrins in blood plasma, urine and red blood cells. Urine and plasma porphyrins show an increase predominantly of uroporphyrin and heptacarboxylporphyrin. Consider erythrocyte zinc protoporphyrin levels, which are significantly increased in hepatoerythropoietic porphyria (HEP), to differentiate hepatoerythropoietic porphyria (HEP) from congenital erythropoietic porphyria (CEP).
Biochemical Characteristics of Hepatoerythropoietic Porphyria 79
- Plasma: Increased uroporphyrin, heptacarboxylporphyrin (~620 nm) (Fluorescence emission peak of diluted plasma at neutral pH, following excitation at 400-410 nm)
- Urine: Increased uroporphyrin, heptacarboxylporphyrin
- Erythrocytes (red blood cells): Increased zinc protoporphyrin
Porphyrin patterns in hepatoerythropoietic porphyria (HEP) are similar to those seen in porphyria cutanea tarda (PCT) with elevation of highly carboxylated porphyrins and isocoproporphyrins. In contrast to porphyria cutanea tarda (PCT), there are markedly increased levels of zinc protoporphyrin in red blood cells in hepatoerythropoietic porphyria (HEP) patients which is due to accumulation of pathway intermediates being metabolized to protoporphyrins.
Hepatoerythropoietic porphyria differential diagnosis
Symptoms of the following disorders can be similar to those of hepatoerythropoietic porphyria (HEP).
Congenital erythropoietic porphyria (CEP) is a rare inherited metabolic disorder resulting from the deficient function of the enzyme uroporphyrinogen III cosynthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Due to the impaired function of UROS (uroporphyrinogen III cosynthase) enzyme, excessive amounts of particular porphyrins accumulate, particularly in the bone marrow, plasma, red blood cells, urine, teeth, and bones. The major symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in the skin cause bullae (blistering) and the fluid-filled sacs rupture, and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. Congenital erythropoietic porphyria (CEP) is inherited as an autosomal recessive genetic disorder. Typically, there is no family history of the disease. Both parents are usually healthy, but each carries a defective gene that they can pass to their children. Affected offspring have two copies of the defective gene, one inherited from each parent.
There are other conditions that may cause signs and symptoms that are similar to those seen in hepatoerythropoietic porphyria (HEP). Such conditions include other cutaneous porphyrias, sporadic porphyria cutanea tarda (i.e., porphyria cutanea tarda type 1 that is not associated with a UROD pathogenic variant), drug-induced photosensitivity, epidermolysis bullosa, various forms of lupus, and solar urticarial.
Sporadic porphyria cutanea tarda also called porphyria cutanea tarda type 1 that is not associated with a UROD genetic mutation is clinically indistinguishable from hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT) and is highly influenced by susceptibility factors associated with porphyria cutanea tarda (PCT). In these cases, the excess porphyrins are produced only in the liver.
The skin histopathologic findings of pseudoporphyria are similar to those of hepatoerythropoietic porphyria (HEP), however, pseudoporphyria is not associated with porphyrin biochemical abnormalities. Medications, chronic kidney failure, and excessive sun exposure or UV radiation have been reported to cause pseudoporphyria 104.
Table 2. Other Types of Porphyria in Hepatoerythropoietic Porphyria Differential Diagnosis
| Disorder | Gene | Mode of inheritance | Skin Lesions | Distinguishing Features / Comment |
|---|---|---|---|---|
| Hereditary coproporphyria (HCP) | CPOX (coproporphyrinogen oxidase) | Autosomal Dominant | Blistering skin lesions closely resembling lesions of congenital erythropoietic porphyria (CEP) |
|
| Variegate porphyria (VP) | PPOX (protoporphyrinogen oxidase) | Autosomal Dominant | Blistering skin lesions are nearly identical to those in hepatoerythropoietic porphyria (HEP). Cutaneous manifestations in hepatoerythropoietic porphyria (HEP) are chronic & blistering like in variegate porphyria (VP) but are usually more severe than those of variegate porphyria (VP), because circulating porphyrin levels in hepatoerythropoietic porphyria (HEP) are usually much higher than in variegate porphyria (VP). |
|
| Familial porphyria cutanea tarda (F-PCT) | UROD (uroporphyrinogen decarboxylase) | Autosomal Dominant | Skin lesions resemble those of hepatoerythropoietic porphyria (HEP) but are less severe & typically begin later, in the 5th or 6th decade of life. |
|
| Congenital erythropoietic porphyria (CEP) |
|
| The skin lesions of congenital erythropoietic porphyria (CEP), like those seen in hepatoerythropoietic porphyria (HEP), appear early in life (i.e., in infancy or childhood) and are severe & mutilating. | In both congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP), ↑ severity is attributed to plasma concentration of porphyrin. Although congenital erythropoietic porphyria (CEP) can be mistaken for hepatoerythropoietic porphyria (HEP), urine porphyrin analysis which shows marked ↑ in uroporphyrin and coproporphyrin type 1 in congenital erythropoietic porphyria (CEP) helps exclude other cutaneous porphyrias. Fecal analysis may be necessary, particularly for persons with late onset. |
Hepatoerythropoietic porphyria treatment
The treatment of hepatoerythropoietic porphyria (HEP) is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with hepatoerythropoietic porphyria (HEP). Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance and/or protection from sunlight will benefit individuals with hepatoerythropoietic porphyria (HEP) and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies (removal of blood) to lower the amount of porphyrins in the liver, which are used to treat individuals with porphyria cutanea tarda (PCT), are generally ineffective in individuals with hepatoerythropoietic porphyria (HEP) since elevated iron levels are not a feature of the disorder 22, 92. Another treatment for porphyria cutanea tarda (PCT), the antimalarial drug hydroxychloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin (rEPO) a synthetic version of the naturally occurring hormone erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with hepatoerythropoietic porphyria (HEP) whose anemia was not associated with increased red cell destruction.
Light Avoidance
Avoiding exposure to sunlight is the most important way to manage symptoms for people with hepatoerythropoietic porphyria (HEP). People with hepatoerythropoietic porphyria (HEP) need to wear protective clothing, and have windows tinted in their cars and homes. Most sunscreens are not effective because they do not block light in the blue-violet range, which is the type of light that triggers reactions in porphyria.
Agents to avoid
Older individuals should avoid known triggering factors: alcohol, oral estrogen, iron overload, smoking, and drugs that induce the cytochrome P450s.
Blister and Wound Care
There is a risk of developing infections in blisters, particularly if they rupture. Prescription antibiotic ointments may be required to manage infections. Care from a dermatologist is recommended.
Hepatoerythropoietic Porphyria Prognosis
Due to the extreme rarity of hepatoerythropoietic porphyria (HEP) with less than 100 reported cases, little is known about the prognosis of hepatoerythropoietic porphyria (HEP) patients 94, 105, 96. The prognosis for hepatoerythropoietic porphyria (HEP) can be complicated by liver involvement, and liver function test must be monitored regularly. However, with proper treatment and management, many patients with hepatoerythropoietic porphyria (HEP) can maintain a good quality of life 106, 107. No increased risk for liver cancer (hepatocellular carcinoma) has been documented in hepatoerythropoietic porphyria (HEP) 79.
Hepatoerythropoietic porphyria (HEP) typically presents early in infancy or early childhood with signs and symptoms such as extreme photosensitivity, recurrent skin blisters (bullae) and skin erosions on sun exposed skin with secondary bacterial infections that may result in severe skin scarring, sclerodermatous change, and deformities of the hand 81. Sun-induced skin redness (erythema) and blistering occurred by age 2 years in 75% of reported cases 81. Sclerodactyly (a condition where the skin on the fingers and/or toes thickens and tightens, making them feel hard and less flexible), osteolysis (active resorption of bone tissue) and shortening of the phalanges, and progressive joint deformities can occur as well 81, 92.
Spontaneous improvement of acute photosensitivity during later childhood, but persistent skin fragility, has been described 108, 109, 110, 111. Other patients have presented in the second or third decade of life with mild skin fragility or photodistributed annular plaques 112, 113, 110, 114.
Other signs and symptoms inlcude abnormal excessive hair (hypertrichosis), red-brown discoloration of teeth (erythrodontia) and pink-to-red urine. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D deficiency. Red blood cells have a shortened life-span with mild or severe hemolytic anemia. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) 94, 81, 92. Current treatment recommendations resemble those for familial porphyria cutanea tarda (F-PCT) 79.
Mild cases of hepatoerythropoietic porphyria (HEP) may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), the most common form of porphyria in humans where the porphyria cutanea tarda (PCT) that may be acquired in 75% to 80% of cases (also known as porphyria cutanea tarda type 1 or sporadic porphyria cutanea tarda) or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance or porphyria cutanea tarda type 2 or familial porphyria cutanea tarda). Hepatoerythropoietic Porphyria (HEP) is the autosomal recessive form of familial porphyria cutanea tarda (F-PCT). Skin photosensitivity is generally much more severe in hepatoerythropoietic porphyria (HEP) than in porphyria cutanea tarda (PCT).
Compared to porphyria cutanea tarda (PCT), the skin features of hepatoerythropoietic porphyria (HEP) typically have earlier onset, increased severity leading to disfigurement, and closer resemblance to those in congenital erythropoietic porphyria (CEP) 85, 92. However, extracutaneous findings including hemolytic anemia are more frequent and severe in congenital erythropoietic porphyria (CEP) than hepatoerythropoietic porphyria (HEP) 85, 92, 113.
In contrast to the autosomal recessive form of variegate porphyria (VP), which is characterized by developmental delay and seizures 115, neurologic abnormalities are not typically associated with hepatoerythropoietic porphyria (HEP) or congenital erythropoietic porphyria (CEP) 85, 116, 117. Nevertheless, developmental delay and seizures have been previously reported in hepatoerythropoietic porphyria (HEP) 118, 88. A 4-year-old boy had delayed speech and language skills then presented with focal seizures and acute left hemiparesis 118. Two young adults, ages 21 and 23 years, with severe hepatoerythropoietic porphyria (HEP) developed generalized seizures and had neuroimaging evidence of cerebral cortical atrophy and punctate calcifications in the frontal lobes, presumably related to hypoxic injury as in other porphyrias 88. These observations, together with recent affected siblings’ developmental delay, support neurologic assessment of hepatoerythropoietic porphyria (HEP) patients in order to better define this possible complication 81.
Cantatore-Francis et al 81 reported 2 sisters with painful polyarticular arthritis, represents a typical (but heretofore unrecognized) inflammatory precedent of joint deformity in hepatoerythropoietic porphyria (HEP) or an idiosyncratic inflammatory process, perhaps triggered by porphyrin deposition together with exposure to ultraviolet light or another environmental insult, remains to be determined.
Anemia was present in more than 50% hepatoerythropoietic porphyria (HEP) patients for whom hematologic status was reported (15/27) 81, but severe anemia requiring transfusions or administration of erythropoietin (EPO) has only been observed in a few individuals 112.
Erythropoietic protoporphyria (Protoporphyria)
Protoporphyrin IX (PPIX) is excreted by the liver into the bile, after which it enters the intestine and is excreted in the feces. Protoporphyrin IX (PPIX) is not soluble in water so is not excreted in the urine. Excess protoporphyrin IX is excreted by the liver into bile, where it becomes insoluble and can crystallize, forming gallstones and obstructing bile flow. Biliary obstruction impairs protoporphyrin IX excretion, resulting in further protoporphyrin IX accumulation and escalating protoporphyrin IX-mediated liver damage. Protoporphyrin IX-mediated liver dysfunction has been reported in over 50% of patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), which may progress to liver failure in up to 5% of patients and a condition caused protoporphyric hepatopathy that sometimes requires liver transplantation 132, 133.
Erythropoietic protoporphyria (EPP) is the third most common type of porphyria, and the most common in childhood. Erythropoietic protoporphyria (EPP) causes very painful photosensitivity and can greatly impair quality of life. Delay in diagnosis is greater than with any other type of porphyria.
Swelling, burning, itching, and redness of the skin may appear during or after exposure to sunlight, including sunlight that passes through window glass. This can cause mild to severe burning pain on sun-exposed areas of the skin. Usually, these symptoms subside in 12 to 24 hours and heal without significant scarring. Blistering and scarring are characteristic of other types of cutaneous porphyria but are unusual in Erythropoietic protoporphyria. Skin manifestations generally begin early childhood and are more severe in the summer.
Erythropoietic protoporphyria (EPP) is caused by mutations in the FECH gene. The FECH gene provides instructions for making an enzyme known as ferrochelatase 134. The ferrochelatase (FECH) enzyme is involved in the production of a molecule called heme. The production of heme is a multi-step process that requires eight different enzymes. Ferrochelatase is responsible for the eighth and final step in this process, in which an iron (Fe2+) atom is inserted into the center of protoporphyrin IX (the product of the seventh step) to form heme. Heme is vital for all of your body’s organs, although it is most abundant in your blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Due to abnormally low levels of ferrochelatase (FECH) enzyme, excessive amounts of protoporphyrin IX (PPIX) build up in the bone marrow, blood plasma, and red blood cells 135. Protoporphyrin IX (PPIX) compounds are formed during the normal process of heme production, but reduced activity of ferrochelatase allows them to accumulate to toxic levels. The excess porphyrins can leak out of developing red blood cells and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins compounds in the skin cause the oversensitivity to sunlight that is characteristic of erythropoietic protoporphyria (EPP). Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
Some patients with symptoms of protoporphyria have a genetic change (gain-of-function variants in exon 11 of ALAS2) in a different gene called ALAS2 gene (delta-aminolevulinic acid synthase-2 gene) and follows an X-linked inheritance pattern 136, 137. When a patient has a genetic change in the ALAS2 gene located on the X chromosome, the condition is referred to as X-linked protoporphyria (XLP) 138. XLP accounts for 2–10% of protoporphyria cases 119. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 137. This version of the enzyme is found only in developing red blood cells called erythroblasts. ALA-synthase enzyme also plays an important role in the production of heme. The production of heme is a multi-step process that requires eight different enzymes. ALA-synthase is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA). The excess delta-aminolevulinic acid (δ-ALA) is converted by other enzymes to compounds called porphyrins. If porphyrins compounds build up in erythroblasts, they can leak out and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP).
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are characterized by a buildup of protoporphyrin in the skin, blood, and liver. It typically presents in early childhood with immediate pain and crying upon exposure to bright sunlight (e.g., babies may cry in the sun or in the car). Erythropoietic protoporphyria signs and symptoms is seasonal in nature with symptoms principally occurring in the spring and summer season.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) signs and symptoms include:
- Painful, non-blistering skin reactions to sunlight or artificial light, most often on the tops of the hands and feet, face and ears. Most individuals with EPP develop acute cutaneous photosensitivity within 30 minutes after exposure to sun or ultraviolet light. Pain can be severe and and can last up to a week after sun exposure 124.
- Itching, burning, or redness of the skin.
- Swelling or edema in the affected areas.
- Persistent redness or inflammation of the skin.
- Pregnancy has been associated with decreased protoporphyrin levels and increased tolerance to sun exposure, but these are inconsistent 139, 140, 141, 142, 143, 144, 145, 146.
- Over the years, the skin on the backs of the hands and cheeks can have some thickening with subtle pitted scarring.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) causes skin pain on exposure to sunlight. There may not be anything to see at the time. Prolonged exposure can result in some redness and swelling, and uncommonly in blistering and crusting.
EPP is a very rare inherited disorder that affects males and females in equal numbers. It is estimated that erythropoietic protoporphyria (EPP) occurs in about 1 in about 75,000 to 1 in 200,000 individuals in Europe, with prevalence figures ranging between 1 in 75,000 (The Netherlands) and 1 in 200,000 (Wales) 147, 148, 127. However, recent genetic evidence has revealed erythropoietic protoporphyria true prevalence is ∼1 in 17,000 149, 150, 151. The number of patients affected by these disorders in the US is unknown. Erythropoietic protoporphyria (EPP) seems that males and females are equally affected 152.
X-linked protoporphyria (XLP) accounts for about 10% of erythropoietic protoporphyria cases in the United States. X-linked protoporphyria (XLP) is more likely to present in males. Females with XLP may or may not have symptoms 127.
The onset of symptoms affecting the skin usually occurs in infancy, with an average of diagnosis at age 4; however, in some cases, onset may not occur until adolescence or rarely even adulthood. A clinical diagnosis of EPP is often made during childhood. The blood needs to be sent for porphyrin analysis in a tube protected from light with aluminium foil.
- The patient’s red blood cells may be noted to fluoresce by ultraviolet microscopy.
- The characteristic change is the elevation of the red cell protoporphyrin.
- Genetic testing for mutations in the ferrochelatase (FECH) gene can be performed.
- A skin biopsy is rarely performed as the skin often appears normal; however, EPP has some characteristic features on histopathology.
Once the diagnosis has been made, regular checks of liver function are required with intermittent liver imaging. Genetic counseling is recommended for affected individuals and their families.
Erythropoietic protoporphyria is a lifelong disease, and repeated phototoxic reactions eventually lead to thickening of the skin and wax-like scarring on the face. In a small number of patients with erythropoietic protoporphyria, the accumulation of protoporphyrins in the liver leads to cirrhosis and liver failure. Onset in adulthood is rare, but an acquired form has been identified, in which clones of cells with mutated ferrochelatase expand in the setting of the myelodysplastic or myeloproliferative syndrome 153.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) diagnosis is made by finding increased levels of the protoporphyrin in the plasma or red blood cells. High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 154. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection. Skin biopsies are not required for the diagnosis of protoporphyrias or any other type of cutaneous porphyria. Genetic testing is useful to confirm the diagnosis.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
For the initial screening for porphyrias with skin symptoms, a plasma fluorescence scan detecting increased plasma porphyrin concentrations is recommended. This approach offers the added benefit of recognizing other forms of porphyrias associated with cutaneous symptoms, that is, porphyria cutanea tarda (PCT), porphyria variegata (VP), hereditary coproporphyria (HC) and congenital erythropoietic porphyria (CEP). Moreover, a negative test result excludes any form of porphyria as the underlying cause of concurrent skin symptoms. In the case of the protoporphyrias, protoporphyrin IX (PPIX) present in the plasma typically causes a positive plasma‐fluorescence scan with a peak at around 633–635 nm 155. The diagnosis of the protoporphyrias is confirmed by the quantification of protoporphyrin IX (PPIX) in red blood cells (erythrocytes), whereby metal‐free PPIX and zinc protoporphyrin IX (ZnPP) are determined separately. A metal‐free PPIX ≥3 times the upper limit of normal (ULN) establishes the diagnosis of protoporphyrias.
To reliably perform a plasma fluorescence scan and the protoporphyrin IX (PPIX) measurement in red blood cells (erythrocytes), the vial of blood (5 mL) needs to be protected from light with an aluminium foil. Exposure of the blood tube to light can lead to photobleaching of PPIX, resulting in a decrease in measured PPIX levels in the sample as compared to the actual levels in the subject. This effect is related to the ability of conjugated double bonds in the porphyrin rings to absorb the energy from visible light 156. To avoid diagnostic delay, plasma porphyrins should be measured at the same time with erythrocyte protoporphyrin 10.
Other test materials used for the diagnosis of other forms of porphyrias are urine and faeces. Urinary porphyrins are not diagnostic in protoporphyrias as, due to its hydrophobicity, excess PPIX is excreted by the biliary route and faeces. Therefore, in the faeces, an increase in PPIX can be present, but the diagnosis requires confirmation by a significant increase of PPIX and zinc protoporphyrin IX (ZnPP)P in the erythrocytes.
Erythropoietic protoporphyria treatment includes the following:
- Avoid sunlight and wear protective clothing.
- Use topical anesthetic creams for the treatment of phototoxic symptoms. Anecdotally, many patients choose to self-treat with ice, cold water, or cold compresses, resulting in minor relief of phototoxic symptoms. However, there is no evidence of benefit with narcotic analgesics, oral or topical corticosteroids, antihistamines, acetaminophen and non-steroidal anti-inflammatory drugs 119.
- Take vitamin D supplements. There is an increased recognition of prevalence of vitamin D deficiency in protoporphyria patients due to lifelong sunlight avoidance. Routine screening for vitamin D deficiency and supplementation as per population guidelines are recommended 157, 158
- Avoid alcohol to prevent additional cause of liver damage 119. Additionally, immunization against hepatitis A and B is recommended.
- Narrowband UVB phototherapy. Narrowband UVB phototherapy does not cause the EPP pain. It is given 3 days a week over 6 weeks in Spring. It increases melanin content causing a tan and induces skin thickening so to provide some level of protection from the sun.
- Scenesse (afamelanotide). Scenesse (afamelanotide) is an alpha-melanocyte stimulating hormone that is given by subcutaneous implantation and works by increasing skin pigmentation which provides protection and improves sun tolerance 128. There is no data on the safety of Scenesse (afamelanotide) during pregnancy so this cannot be recommended for pregnant women.
- In patients undergoing surgery of prolonged duration, such as liver transplantation, light filters that limit transmission of the wavelengths 340–470 nm (i.e., acrylate yellow filter) are recommended 159, 160. No specific anesthetic agents or other medications are contraindicated in protoporphyria.
- Due to insufficient data related to efficacy, the following therapies are not recommended for the prevention of phototoxic symptoms: beta-carotene, cysteine, cimetidine, isoniazid, warfarin, quinacrine, oral zinc, N-acetylcysteine, vitamin C, omega-3 fatty acids, oral adenosine monophosphate, canthaxanthine, terfenadine, inosine, dithiothreitol (DTT) and glycerol, pyridoxine, and hydroxyethylrutosides 161, 162, 163, 119. Although several previous studies have investigated beta carotene in EPP, the evidence shows unclear or no benefit 119. Dersimelagon is a synthetic, orally administered, small molecule agonist of melanocortin-1 receptor (MC1R) being tested for the prevention of protoporphyria phototoxicity. A positive Phase 2 clinical trial has been completed showing promising results, and a Phase 3 study is ongoing 164. There are no approved therapies for children patients.
Figure 15. Erythropoietic protoporphyria
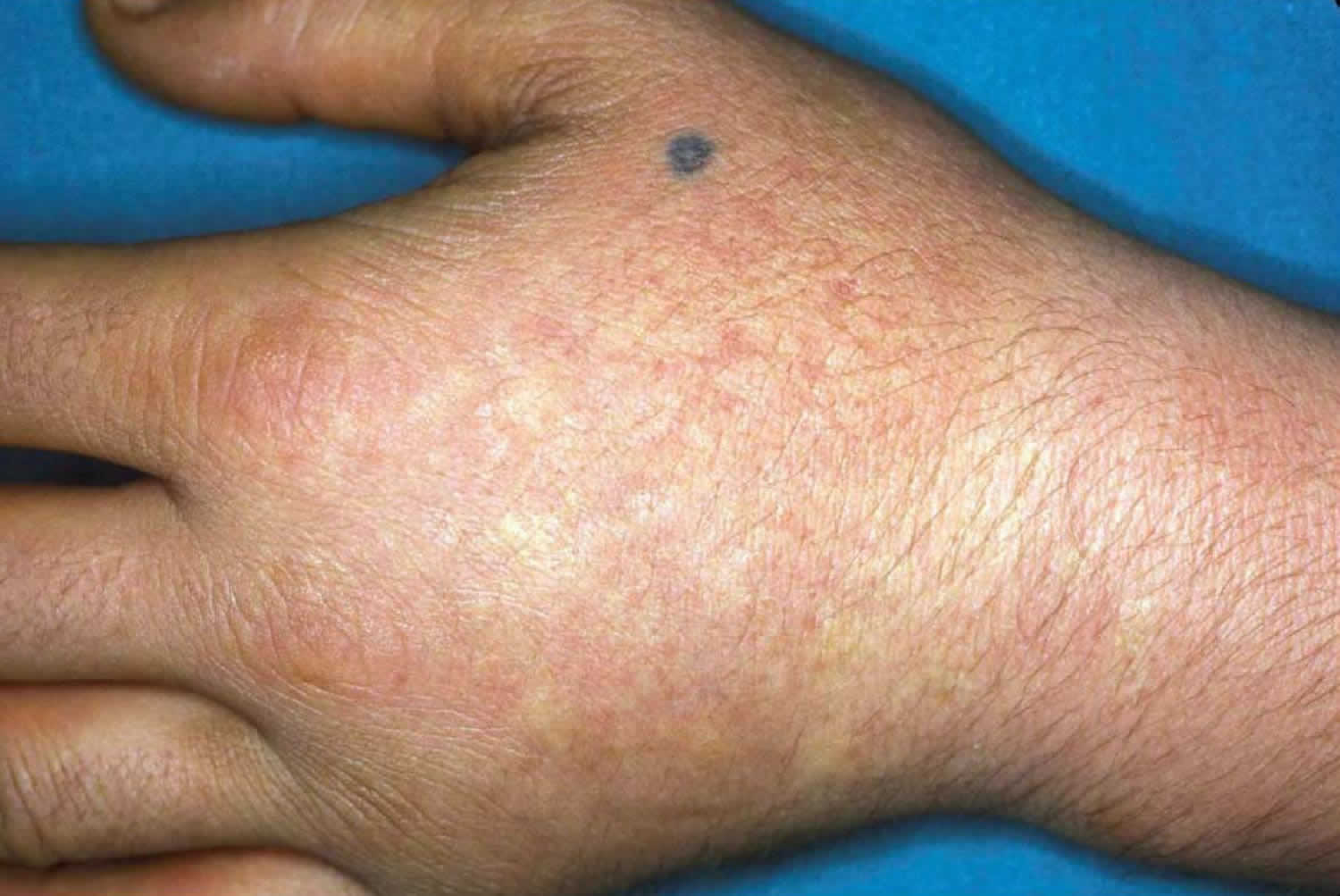
Footnote: Acute photosensitivity reaction in erythropoietic protoporphyria (EPP).
[Source 124 ]Figure 16. Erythropoietic protoporphyria
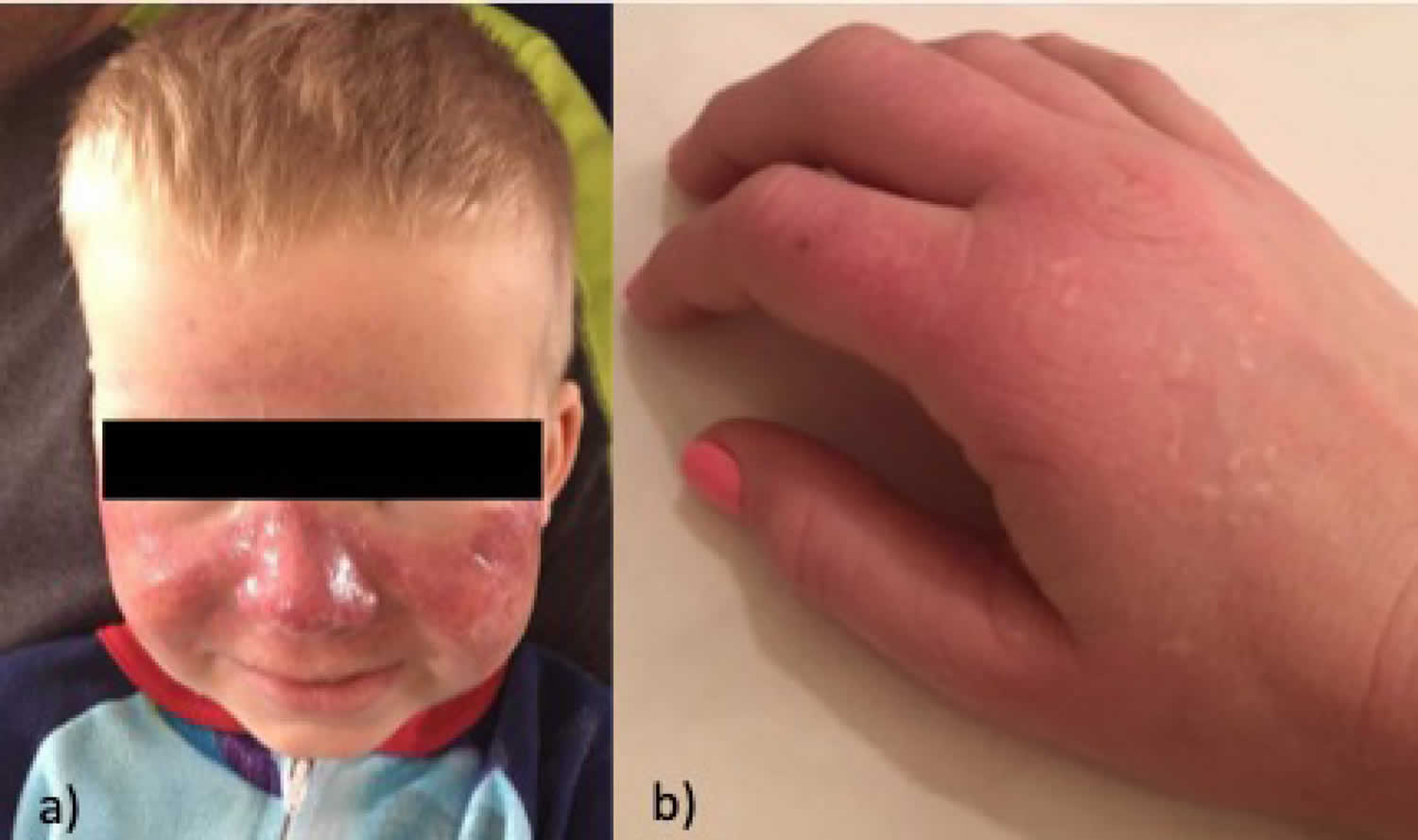
Footnotes: a) Child with extensive edema of the face with erythema and petechiae; b) Adult patient with skin redness (erythema) and skin swelling (edema) during phototoxic episode, with hypopigmented scars and skin thickening present
[Source 119 ]Figure 17. Chronic erythropoietic protoporphyria skin lesions
Erythropoietic protoporphyria signs and symptoms
The most common symptom of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with pain, itching, and/or burning of the skin occurring after exposure to sunlight and occasionally to fluorescent light. Some patients may also be sensitive to some types of artificial light such as fluorescent lights. When the skin is exposed to sun, people with erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) first develop tingling, itching, and/or burning of the skin 127. These symptoms serve as warning signs as longer exposure can result in severe pain. Affected individuals may also have an abnormal accumulation of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema) 127. In rare cases, affected areas of the skin may develop sac-like lesions (vesicles or bullae) and scar if exposure to sunlight is prolonged 127. However, scarring and/or discoloring of the skin (hyperpigmentation) is uncommon and rarely severe 127. The affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the nails. The severity and degree of photosensitivity is different from case to case. Photosensitivity is often seen during infancy; however, in some cases, it may not occur until adolescence or adulthood.
The severity and degree of symptoms is different from case to case. Some patients may only be able to tolerate a few minutes of sun exposure while others may be able to tolerate longer sun exposure without symptoms. The amount of sun tolerated may also be different based on weather conditions. Symptoms are often seen during infancy; however, in some cases, it may not occur until adolescence or rarely in adulthood.
Symptoms usually start in childhood but diagnosis is often delayed since skin blistering is not common and because the porphyrins are insoluble, they cannot be detected on urine analysis. The diagnosis is made upon finding increased levels of the protoporphyrin IX (PPIX) in the plasma or red blood cells.
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, gallstones can cause obstruction and/or inflammation of the gallbladder (cholecystitis).
In some affected individuals, the flow of bile through the gallbladder and bile ducts (biliary system) may be interrupted (cholestasis) causing gallstones (cholelithiasis) to form. In turn, such stones can cause obstruction and/or inflammation of the gallbladder (cholecystitis).
Rarely, affected individuals may also develop liver damage that, in very severe cases, may lead to liver failure requiring liver transplantation. As liver transplantation does not cure erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP), a bone marrow transplant following liver transplant may be necessary in some cases 127.
Patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) may also have mild anemia (low blood counts). In many cases, this may be due to low iron stores (iron deficiency). They may also have high levels of liver enzymes on blood tests.
Liver involvement
Patients with erythropoietic protoporphyria (EPP) may also have liver disease that require specialist medical treatment and possibly liver transplantation. Liver disease develops in association with erythropoietic protoporphyria (EPP) in 1 to 4% of cases 148, 152, with usual features of visceral enlargement and portal hypertension. Liver disease in protoporphyria is related to the excess protoporphyrin cleared by the entero-hepatic circulation leading to paracrystalin protoporphyrin deposition in liver cells (hepatocytes) and precipitation in the biliary canaliculiy. The percentage of patients who will develop liver disease is not established, nor specific factors that may influence its development. In EPP, the degree of severity of the liver disease is variable. Liver disease in EPP may include: gallstones (cholelithiasis) with possible obstructive episodes and chronic liver disease evolving to rapid acute liver failure 165, 166.
The incidence of gallstones (cholelithiasis) is frequent in about 20% of EPP patients EPP 124. Gallstones with high protoporphyrin content are generated due to the accumulation of insoluble protoporphyrin and increased biliary protoporphyrin concentration 167.
It is not possible to predict whether or not acute liver failure will occur. Studies have revealed that an increase in coproporphyrin urinary excretion, together with a change in isomer predominance from isomer III to isomer I, and increasing levels of protoporphyrinaemia may precede this complication 168.
Progression of protoporphyric liver deterioration leads to splenomegaly, splenic sequestration of erythrocytes with haemolysis (which increases erythropoiesis) and protoporphyrin generation ending in fulminant liver failure 165, 168, 169.
Erythropoietic protoporphyria complications
The main serious complication associated with EPP is protoporphyrin-related liver disease, which may be fatal 126. Severe liver disease affects 2%–5% of protoporphyia patients 170. Liver injury occurs from the crystallization of protoporphyrin in the bile ducts 171. This obstruction of the biliary system further increases plasma protoporphyrin, which is typically excreted in the bile, escalating the protoporphyrin-mediated liver damage that may progress rapidly 171. Previous studies indicate that higher protoporphyrin levels may be associated with an increased risk of liver disease or progression 171. Genetic sequencing to identify EPP patients with two pathogenic FECH variants other than c.315-48T>C may be considered, as these patients may be more likely to progress to liver failure 172. Therefore, liver function tests should be performed at the time of diagnosis and at least yearly thereafter 173, 174, 175. This may allow for early identification, evaluation for additional factors contributing to liver disease, and medical management.
The extent to which alcohol use worsens protoporphyria-related liver disease or whether a safe limit alcohol use exists remains unclear 119. Avoidance of excessive alcohol intake is recommended for all people with EPP or XLP 119. Furthermore, immunization against hepatitis A and B is recommended. Although protoporphyria patients are at an increased risk for gallstones (cholelithiasis), in asymptomatic patients with normal liver chemistries, a screening ultrasound is not recommended 119. Surgery to remove your gallbladder (cholecystectomy) is not recommended for asymptomatic gallstones (cholelithiasis).
In the later stages of liver disease, the patient may develop peripheral nerve damage (neuropathy), which mimics the peripheral neuropathy of acute porphyria and may lead to breathing failure 126. On repeated exposure to sunlight, the skin over the face, dorsum of hands (knuckles), can become thickened or lichenified along with loss of lunulae of fingernails. Due to regular sunlight avoidance, patients with EPP are more prone to develop a vitamin-D deficiency, which can lead to osteoporosis 138.
Erythropoietic protoporphyria causes
Erythropoietic protoporphyria (EPP) is caused by mutations in the FECH gene. The vast majority of patients with EPP are compound heterozygotes for both a rare pathogenic FECH variant and a common low expression FECH allele that is present in ~10% of the Caucasian population (c.315-48T>C), a combination that results in ~30% of normal enzyme activity 176, 177. Homozygosity for FECH c.315-48T>C does not cause EPP 178. The FECH gene provides instructions for making an enzyme known as ferrochelatase 134. The ferrochelatase (FECH) enzyme is involved in the production of a molecule called heme. The production of heme is a multi-step process that requires eight different enzymes. Ferrochelatase is responsible for the eighth and final step in this process, in which an iron (Fe2+) atom is inserted into the center of protoporphyrin IX (the product of the seventh step) to form heme. Heme is vital for all of your body’s organs, although it is most abundant in your blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Due to abnormally low levels of ferrochelatase (FECH) enzyme, excessive amounts of protoporphyrin build up in the bone marrow, blood plasma, and red blood cells. Porphyrins compounds are formed during the normal process of heme production, but reduced activity of ferrochelatase allows them to accumulate to toxic levels. The excess porphyrins can leak out of developing red blood cells and be transported through the bloodstream to your skin and other tissues such as your liver. High levels of porphyrins compounds in the skin cause the oversensitivity to sunlight that causes severe pain and inflammation resulting in symptoms of EPP. Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
Some patients with symptoms of protoporphyria have a genetic change (gain-of-function variants in exon 11 of ALAS2) in a different gene called ALAS2, a gene located on the X chromosome and follows an X-linked inheritance pattern 136. When a patient has a genetic change in the ALAS2 gene, the condition is referred to as X-linked protoporphyria (XLP). XLP accounts for 2–10% of protoporphyria cases 119. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 137. This version of the enzyme is found only in developing red blood cells called erythroblasts. ALA-synthase enzyme also plays an important role in the production of heme. The production of heme is a multi-step process that requires eight different enzymes. ALA-synthase is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA). The excess delta-aminolevulinic acid (δ-ALA) is converted by other enzymes to compounds called porphyrins. If porphyrins compounds build up in erythroblasts, they can leak out and be transported through the bloodstream to your skin and other tissues. High levels of porphyrins in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP).
About 4 to 6% of patients with the symptoms of erythropoietic protoporphyria (EPP) and elevated red blood cell protoporphyrin IX (PPIX) levels will not have mutations in ferrochelatase (FECH) or 5-aminolevulinate synthase 2 (ALAS2) 179, 180.
Recently, an autosomal dominant mutation in human CLPX, a modulator of heme biosynthesis, was found to result in the accumulation of delta-aminolevulinate synthase (ALAS) and protoporphyrin IX (PPIX) and symptoms of protoporphyria in an affected family 181. Acquired somatic ferrochelatase (FECH) mutations have been identified in a small number of patients in whom erythropoietic protoporphyria (EPP) has developed after the age of 40 years in association with myelodysplasia or myeloproliferative disorder 182, 183. A case of late-onset erythropoietic protoporphyria (EPP) with myelodysplastic syndrome has also been reported in a patient who had the homozygous IVS3–48T>C polymorphism in the ferrochelatase (FECH) gene 184. Late onset X-linked protoporphyria (XLP) has also been reported in a case of early myelodysplastic syndrome with somatic mosaicism in the bone marrow 185.
Erythropoietic protoporphyria inheritance pattern
Erythropoietic protoporphyria (EPP) is inherited in an autosomal recessive manner 2, 127. Everyone has two copies of the FECH gene, one inherited from the mother and one from the father. Most individuals with EPP have a different gene mutation on each copy of the FECH genes. On one copy, the change, called a mutation, has stopped this copy of the FECH gene from working properly. On the other copy, there is a small change called a “low-expression allele” or a polymorphism. This alteration still affects the way the FECH gene works; it produces less ferrochelatase enzyme than normal. This small change is common in the general population, with up to 10% of Caucasians with one copy of this change. This alteration will not cause EPP by itself, and people who have the alteration on each copy of the FECH gene will NOT develop EPP. But when someone inherits the small alteration from one parent and a mutation from the other, they will develop EPP, because there will not be enough ferrochelatase (FECH) enzyme made. Most patients with EPP have the low-expression alteration on one copy of the FECH gene and a mutation on the other copy. The risk for patients with EPP to have a child who also has the condition depends on the genetic changes in their partner.
Erythropoietic protoporphyria is a rare disorder inherited as an autosomal dominant genetic trait with poor penetrance. Human traits, including the classic genetic diseases, are the product of the interaction of two genes, one received from the father and one from the mother.
In dominant disorders, a single copy of the disease gene (received from either the mother or father) will be expressed “dominating” the other normal gene and resulting in the appearance of the disease. The risk of transmitting the disorder from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child. The risk is the same for each pregnancy.
The symptoms of erythropoietic protoporphyria develop due to excessive levels of a chemical called protoporphyrin that accumulates in certain tissues of the body (i.e., the plasma, red blood cells, and the liver). Excessive protoporphyrin levels occur as the result of abnormally low levels of the enzyme ferrochelatase (FECH).
There are several different allelic variants of erythropoietic protoporphyria. An allele is any of a series of two or more genes that may occupy the same position (locus) on a specific chromosome. Symptoms of these allelic variants of erythropoietic protoporphyria are predominantly the same; however, one type may be inherited as an autosomal recessive genetic trait.
The gene that is responsible for regulating the production of the enzyme ferrochelatase (FECH) has been located on the long arm of chromosome 18 (18q21.3). Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males, and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q”.
Some people who have inherited this defective gene may have slightly elevated levels of protoporphyrin in the body but will not exhibit the symptoms of erythropoietic protoporphyria.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 18. Erythropoietic protoporphyria (EPP) autosomal recessive inheritance pattern
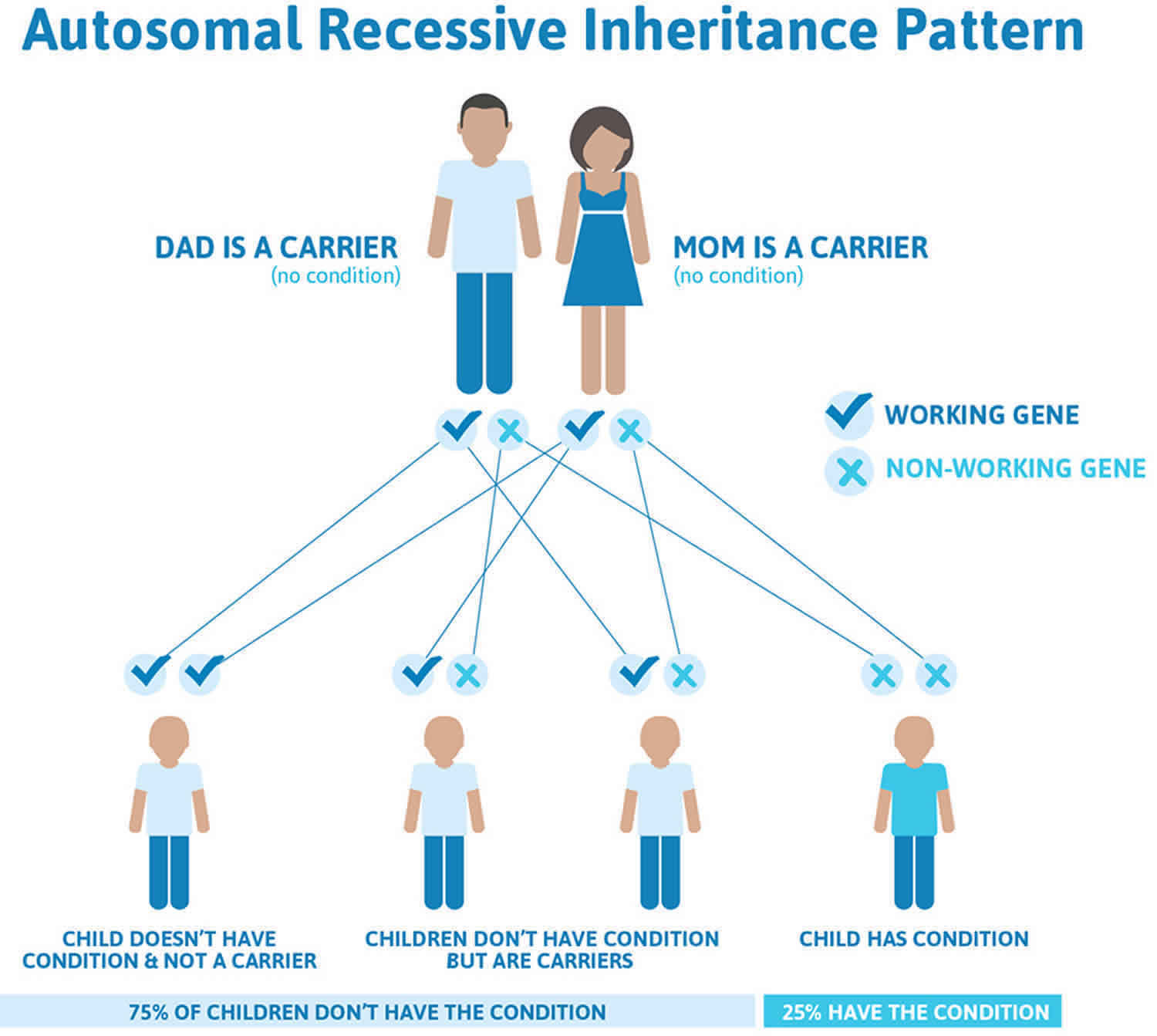
Erythropoietic protoporphyria diagnosis
Erythropoietic protoporphyria (EPP) should be suspected in anyone with non-blistering photosensitivity especially when it is prolonged and beginning in childhood. It is easy to make a diagnosis, or rule it out, once it is suspected. The diagnosis of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) may be made by a thorough clinical evaluation, and specialized laboratory tests. Total red blood cell (erythrocyte) protoporphyrin concentration, including proportions of metal-free and zinc-bound protoporphyrin, is the recommended test for the diagnosis of protoporphyria 186, 187, 188. High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 154. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection. Genetic testing is useful to confirm the diagnosis.
The diagnosis of erythropoietic protoporphyria is established by finding an abnormally high level of total red blood cell (erythrocyte) protoporphyrin, and showing that this increase is mostly free protoporphyrin rather than zinc protoporphyrin. There is considerable confusion about which test to order. Sometimes laboratories have measured only zinc protoporphyrin and reported results incorrectly as “protoporphyrin” or “free erythrocyte protoporphyrin (FEP)”.
Porphyrins are almost always elevated in plasma in erythropoietic protoporphyria, but may be normal in mild cases. Fecal porphyrins may be normal or increased.
An experienced biochemical laboratory can usually distinguish between patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), because erythropoietic protoporphyria (EPP) have much less zinc protoporphyrin in their erythrocytes. This can be explained because in the bone marrow the enzyme ferrochelatase not only normally makes heme (iron protoporphyrin) from protoporphyrin and iron, but can also make zinc protoporphyrin, especially when excess protoporphyrin is present or iron is deficient. However, this does not replace DNA studies or genetic testing.
Genetic testing of the FECH and ALAS2 genes is useful to confirm the diagnosis and identify if it is erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 176. This information is useful for genetic counseling and testing family members as both are inherited in a different manner. Genetic counseling is recommended for couples with a personal or family history of protoporphyria who are planning to have children.
Erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) are usually diagnosed during infancy or early childhood, due to characteristic symptoms, and by testing the red blood cells (erythrocytes) for increased levels of protoporphyrin IX (PPIX). The blood needs to be sent for porphyrin analysis in a tube protected from light with aluminium foil. Exposure of the blood tube to light can lead to photobleaching of protoporphyrin IX (PPIX), resulting in a decrease in measured protoporphyrin IX (PPIX) levels in the sample as compared to the actual levels in the subject. This effect is related to the ability of conjugated double bonds in the porphyrin rings to absorb the energy from visible light 156. To avoid diagnostic delay, plasma porphyrins should be measured at the same time with erythrocyte protoporphyrin 10.
The normal total protoporphyrin level in red blood cells (erythrocytes) is 80 mcg/dL, but in a patient with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), protoporphyrin level in red blood cells (erythrocytes) is elevated up to 300 to 8000 mcg/dL 126. There is an increased percentage of red blood cell (erythrocyte) metal-free protoporphyrin rather than zinc protoporphyrin 126. In patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP), the urinary porphyrin levels are normal.
Rarely, erythropoietic protoporphyria (EPP) develops in adults in the presence of a bone marrow disorder such as polycythemia vera, and is due to expansion of a clone of red blood cell precursors in the marrow that is deficient in ferrochelase.
DNA studies or genetic testings are important for confirming the diagnosis of erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) and for genetic counseling. This should be completed first in a person known to have the disease, and the information about the mutations in that individual used to guide testing of family members.
When erythropoietic protoporphyria (EPP) is due to a FECH mutation the inheritance is described as autosomal recessive. It is most common to find that one severe mutation is inherited from one parent and another weak mutation inherited from the other parent. The weak mutation is quite common in normal Caucasians, rare in Blacks and even more common in Japanese and Chinese populations. This mutation is sometime referred to as “hypomorphic” because it results in formation of a less than normal amount of ferrochelatase. But is does not cause Erythropoietic protoporphyria unless it is paired with a severe mutation. The severe mutation is characteristic for an Erythropoietic protoporphyria family and is present in all affected individuals. “Carriers” of the severe mutation are not affected because they do not have the weak mutation. Affected individuals and unaffected carriers can transmit the severe mutation to the next generation. Some of their children will have Erythropoietic protoporphyria if the other parent has a copy of the weak mutation. Rarely, the weak mutation is absent in an Erythropoietic protoporphyria family and two severe mutations are found, with at least one producing some ferrochelatase.
In X-linked protoporphyria (XLP), mutations of the ALAS2 gene, which is found on the X chromosome, causes an increase in the production of the enzyme ALAS2 in the bone marrow. Several of these “gain of function” mutations have been described in different X-linked protoporphyria families. In X-linked protoporphyria protoporphyrin production exceeds that needed for heme and hemoglobin formation. Like hemophilia and other X linked genetic diseases, X-linked protoporphyria is more common in men. Women have two X chromosomes and are usually not affected because they have a normal as well as a mutated ALAS2 gene. Men have only one X chromosome and will be affected if they inherit an ALAS2 mutation. Women with an ALAS2 mutation will, on average, pass that mutation to half of their daughters (who will usually be unaffected carriers) and to half of their sons (who will be affected).
Figure 19. Erythropoietic protoporphyria diagnostic algorithm
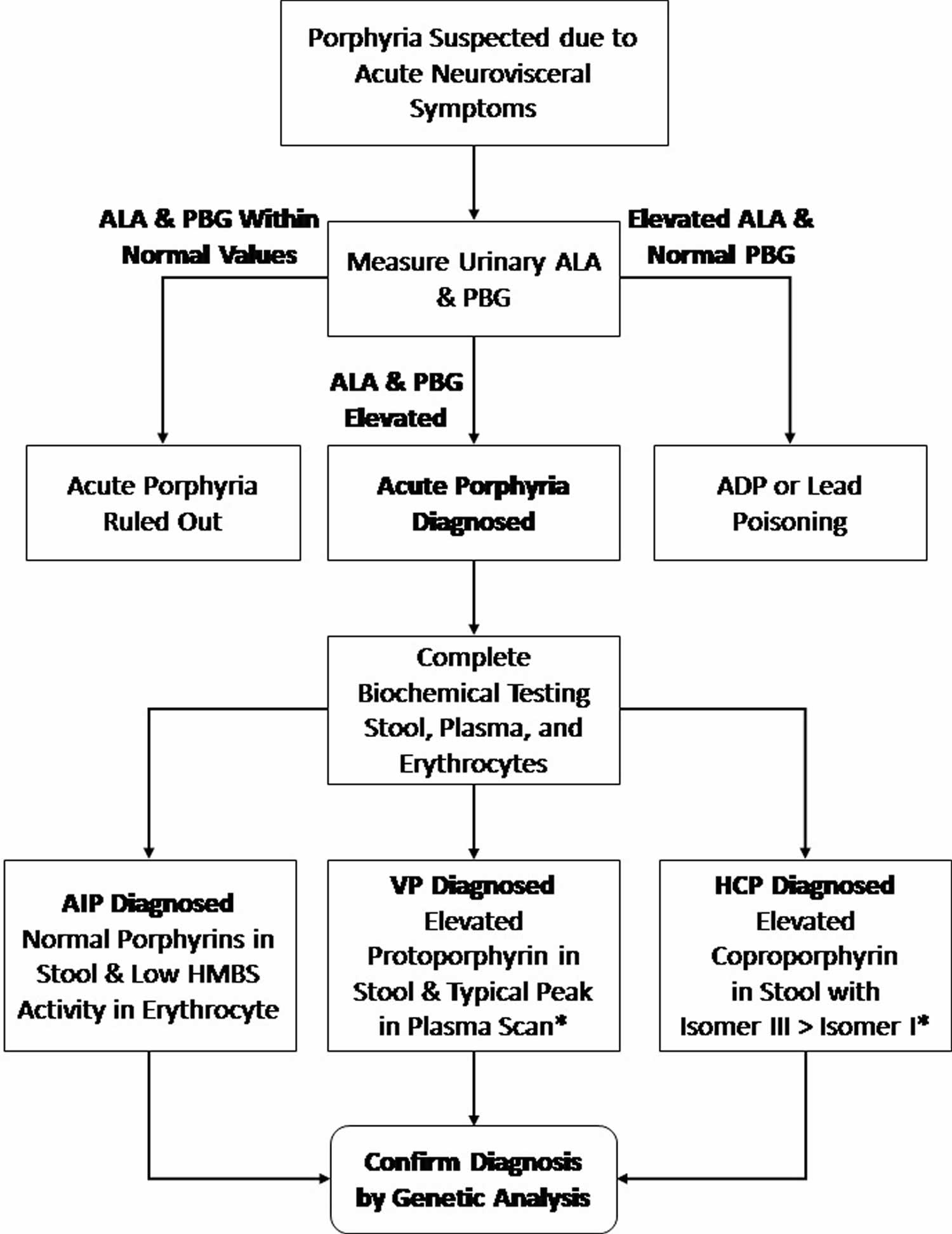
Abbreviations: ADP = aminolevulinic acid dehydratase porphyria; AIP = acute intermittent porphyria; ALA = aminolevulinic acid; HCP = hereditary coproporphyria; HMBS = hydroxymethylbilane synthase; PBG = porphobilinogen; VP = variegate porphyria.
[Source 10 ]Pitfalls of testing
The biochemical diagnosis of protoporphyrias is based on the measurement of red blood cell (erythrocyte) total and metal-free protoporphyrin 121. The latter comprises 85% to 100% of total red blood cell (erythrocyte) protoporphyrin in EPP, and 50% to 85% in XLP 189, 154. Zinc protoporphyrin predominates in other conditions that increase red blood cell (erythrocyte) protoporphyrin. Plasma and fecal porphyrin levels can be variably increased, and urine porphyrin levels are normal. Genetic testing is recommended to distinguish between EPP and XLP and to confirm the diagnosis 189.
Measurement of red blood cell (erythrocyte) protoporphyrin levels is fraught with a lack of standardized nomenclature and methodology 190. Some large commercial laboratories measure protoporphyrin by hematofluorometry, a method that only measures zinc protoporphyrin and that was developed to screen for lead poisoning and iron deficiency. Hematofluorometers report the molar ratio of zinc protoporphyrin to heme by fluorescence but do not measure total or metal-free protoporphyrin 154. Nevertheless, these major laboratories report “free erythrocyte protoporphyrin” (FEP) levels that, in actuality, reflect only zinc protoporphyrin, which may not be increased in EPP. This inappropriate method and inaccurate terminology propagates confusion that can lead to missed diagnoses of protoporphyria 190. Accurate testing reports should only refer to erythrocyte total protoporphyrin, zinc protoporphyrin, and metal-free protoporphyrin, and comment whether the results are consistent with protoporphyria. The term FEP became unclear after the discovery of zinc protoporphyrin in erythrocytes in the 1970s and should no longer be used 121.
High performance liquid chromatography or extraction methods that measure total, metal-free, and zinc protoporphyrin are recommended for the diagnosis of protoporphyria 154. Current lists of laboratories that perform such testing can be found on websites for the United Porphyrias Association, the European Porphyria Network, and the American Porphyria Foundation. These laboratories can also give advice regarding the optimal laboratory testing or differential diagnosis of biochemical abnormalities. As sample materials and pre‐analytical specifications depend on the specific diagnostic methods, it is advisable to contact the respective laboratory prior to sample collection.
Erythropoietic protoporphyria differential diagnosis
Erythropoietic protoporphyria differential diagnosis include 126:
- Phototoxic drug reaction: Phototoxic drug reaction is a non-immunologic skin reaction that appears acutely within minutes to hours on sun-exposed skin after taking photosensitising medications. There must be a history of the introduction of any new drug. Phototoxic skin damage begins when the drug or its metabolite within the skin absorbs ultraviolet radiation (UVR) or visible light. Patients experience painful reddish skin immediately after sun exposure 191, 192.
- Hydroa vacciniforme: Hydroa vacciniforme is one of the rarest forms of photosensitivity dermatoses. Hydroa vacciniforme affects sun-exposed skin and is characterized by recurrent fluid-filled blisters (‘hydroa’) over sun-exposed sites that heal with pox-like (‘vacciniform’) scars 193.
- Solar urticaria: Solar urticaria is a condition in which exposure to sunlight or an artificial light source emitting ultraviolet radiation causes urticaria 194. Like EPP, symptoms often develop within minutes. Symptoms of solar urticaria are often itchy rather than painful. The reaction may subside within a few minutes or it may persist for up to an hour or more where it can become very disabling. The cause of solar urticaria is not clearly defined but may be due to an antigen-antibody reaction 194. It seems that a chemical created in the body (a photoallergen) reacts with UV radiation to cause an allergic reaction that manifests as urticaria.
- Polymorphic light eruption (PMLE): Polymorphic light eruption also called a sun allergy or sun poisoning. Polymorphic light eruption is a common seasonal, acquired, idiopathic photodermatosis occurring in spring and early summer that typically occurs during the first three decades of life 195. Symptoms occur in sun-exposed areas. Patients present with discrete lesions such as pruritic papules, vesicles, or plaques on sun-exposed areas.
- Discoid lupus erythematosus: It presents as scaly erythematous plaques on sun-exposed areas.
- Sunburn: It is a transient inflammatory skin response to ultraviolet radiation from sunlight or artificial sources. Sunburn can occur in individuals without an underlying dermatologic condition, with sensitivity depending on the degree of skin pigmentation and hair and eye color 196.
Erythropoietic protoporphyria treatment
There is no cure for erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). Lifelong photosensitivity is the main problem. To reduce pain, avoid unnecessary exposure to sunlight will be of benefit to individuals with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). The use of sun protective clothing such as long sleeves, wide-brimmed hats, and sunglasses will also benefit individuals with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP). Tanning creams which increase skin pigmentation or sunscreens which contain physical reflecting agents such as zinc oxide or titanium dioxide that reflect visible light may be beneficial to some patients. Individuals with EPP and XLP may also benefit from window tinting or using films to cover the windows in their car or house. Other sources of light may also cause symptoms, including fluorescent and halogen lights. Protect the skin from exposure to operating visible light and ultraviolet A (UVA) light during a surgical procedure. Light filters that limit transmission of the wavelengths 340–470 nm (i.e., acrylate yellow filter) are recommended and should be used in operating rooms to protect the patient, especially in case of long surgical procedures like liver transplantation in a patient with erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 159, 160, 197. No specific anesthetic agents or other medications are contraindicated in protoporphyria.
Avoidance of alcohol is important to reduce the risk of liver damage and liver failure. A drug called Prevalite (cholestyramine) or activated charcoal maybe prescribed to interrupt the circulation of protoporphyrin through the liver and intestines in patients with liver disease.
Once the pain has started, pain relief can be difficult to achieve. Most patients immerse the affected areas in cold water or use ice packs. Topical anesthetic creams can be helpful.
In 2019, the Food and Drug Administration (FDA) approved Scenesse (afamelanotide) for the treatment of adult patients with EPP. Scenesse (afamelanotide) is an alpha-melanocyte stimulating hormone that is given by subcutaneous implantation and works by increasing skin pigmentation which provides protection and improves sun tolerance 128. Afamelanotide binds to the melanocortin-1 receptor (MC1R) and increases eumelanin production from dermal cells and melanocytes, thus increasing pigmentation and providing photoprotection. Melanin is additionally a strong antioxidant thought to inactivate the reactive oxygen species (ROS) produced during phototoxic reactions 198. In the setting of erythropoietic protoporphyria (EPP), melanin production may provide defense against oxidative stress by neutralizing free radicals and the reactive oxygen species produced, thus helping decrease symptom severity 199 Scenesse (afamelanotide) was available in Europe for a period of time before its approval in the United States. There is no data on the safety of Scenesse (afamelanotide) during pregnancy so this cannot be recommended for pregnant women.
Iron supplementation should be avoided unless severe iron deficiency is present — as iron can increase photosensitivity in EPP. Both clinical improvement and increased photosensitivity have been reported during iron replacement therapy 200, 201, 202, 203, 204, 205, 206, 207, 208, 209.
- Iron deficiency in ferrochelatase-deficient EPP: If symptomatic from iron deficiency and/or have hemoglobin levels <10g/dL and ferritin <10ug/L. Iron supplementation to a goal ferritin of 50–100ug/L
- Iron deficiency in XLP: If any iron deficiency: Oral iron to a goal ferritin of 50–100 ug/L
Trials of treatment for erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) have been difficult to assess. Effective treatment should reduce pain and increase time outdoors without pain.
- Narrowband UVB phototherapy does not cause the EPP pain. It is given 3 days a week over 6 weeks in Spring. It increases melanin content (causing a tan) and induces skin thickening so to provide some level of protection from the sun.
- Oral antioxidants such as beta-carotene, Polypodium leucotomas extract, warfarin and N-acetyl cysteine have been used but studies showing effectiveness are lacking. In EPP, a high potency form of oral Lumitene (oral beta-carotene) 120-180 mg/dL has been used to improve an affected individual’s tolerance of sunlight. The dose of Lumitene depends on age, ranging from two to ten 30-mg capsules per day and usually started six to eight weeks before summer. While some patients report improvement, recent studies show that there is no data to support the benefit of this treatment 210.
In addition, individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
Liver transplantation has been performed as a life-saving measure in patients with EPP and XLP related liver failure. Bone marrow transplant can also be performed after liver transplant to prevent further damage to the liver.
EPP and XLP patients should also receive vaccination against hepatitis A and B to prevent other causes of liver damage.
Patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Other treatment is symptomatic and supportive.
1. Sunlight protection
Protection from sunlight is the mainstay of management of erythropoietic protoporphyria, and this is necessary throughout life. Disease severity and porphyrin levels in erythrocytes and plasma probably remain high and relatively constant throughout life in Erythropoietic protoporphyria. However, this has been little studied and more longitudinal observations are needed. Life style, employment, travel and recreation require adjustment in order to avoid painful reactions to sunlight and even from exposure to fluorescent lighting. For these reasons Erythropoietic protoporphyria can substantially affect quality of life.
Protective clothing, including broad-brimmed hats, long sleeves, gloves and trousers (rather than shorts), is beneficial. Several manufacturers specialize on clothing made of closely woven fabrics for people with photosensitivity.
2. Other considerations
In an occasional patient, protoporphyrin causes liver problems, so monitoring liver function is important. Erythropoietic protoporphyria patients should also not use any drug or anesthetic which causes cholestasis (slowing down bile flow), and should also avoid alcohol. Women should avoid medications containing estrogen (birth-control pills, hormone replacement therapy), and men should avoid testosterone supplements, as these substances can also have deleterious effects on the liver of a person with Erythropoietic protoporphyria.
Because erythropoietic protoporphyria is a rare condition, most physicians are not knowledgeable about it. A Medic Alert bracelet with instructions to contact a specialist if needed is a worthwhile precaution.
Yearly monitoring. Testing to include erythrocyte total protoporphyrin, plasma porphyrin, complete blood counts, ferritin and liver function tests should be done yearly. Porphyry levels are expected to be stable and liver tests to remain normal. Erythropoietic protoporphyria patients may have evidence of iron deficiency, and an iron supplement may be advisable if the serum ferritin is below about 20 ng/mL.
Vitamin D. Because they avoid sunlight, Erythropoietic protoporphyria patients are likely to be deficient in vitamin D. A vitamin D supplement with calcium is recommended for bone health.
Liver protection. It is important to avoid other causes of liver disease that might promote the development of liver complications from erythropoietic protoporphyria. Patients should avoid alcohol and other substances that might damage the liver, including many herbal preparations, and be vaccinated for hepatitis A and B.
Surgical lights. Strong operating room lights can cause photosensitivity of the skin and even surfaces of internal organs. Flexible membrane filters, such as CL5-200-X from Madico Co., are available to cover surgical lights and offer some protection. This is especially important in erythropoietic protoporphyria patients with liver failure, which causes even greater increases in protoporphyrin levels and photosensitivity.
Drugs. Drugs that are harmful in other porphyrias are not known to make erythropoietic protoporphyria worse, but are best avoided as a precaution. This may include estrogens and other drugs that might reduce bile formation. A short course of a non-steroidal anti-inflammatory drug can provide some pain relief after an episode of photosensitivity, but can cause ulcerations of the digestive track especially with prolonged use.
Laser treatment. According to Dr. Roth, laser treatments for hair removal or eye surgery have not been a problem in erythropoietic protoporphyria people. But the doctor should be made aware of the diagnosis, and that laser output between 400 and 650 nanometers might be harmful. Before hair removal treatment, the doctor may irradiate a small area of the skin to be treated for the length of time it will take to do the hair removal to ascertain if the patient would react within the period of time that a reaction to sunlight would be expected in that patient.
Children with erythropoietic protoporphyria. Avoiding sunlight can be difficult for children with erythropoietic protoporphyria who have less sunlight tolerance than their friends.
Liver disease treatment
2%–5% of affected individuals develop severe liver complications that are difficult to treat, often requiring liver transplantation 170, 145. Liver complications may be accompanied by motor neuropathy.
Rapidly progressive and severe liver disease can be treated with a regimen that includes intravenous hemin (to reduce plasma porphyrin level), plasmapheresis, ursodeoxycholic acid, cholestyramine, vitamin-E (400IU), and correction of anemia 126, 211. Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement 212.
Liver transplantation is the treatment choice for severe protoporphyric liver damage or for patients who develop liver cirrhosis as a life-saving measure in individuals with severe protoporphyric liver disease 213, 214, 215. However, liver transplant recipients may experience a recurrence of protoporphyric liver disease in the transplanted liver. Combined bone marrow and liver transplantation is indicated in patients with liver failure to prevent future damage to the allografts 216.
Bone marrow transplant is a newer treatment that may be done in conjunction with liver transplantation to prevent the recurrence of EPP, and reports show that it may be able to reverse or treat both photosensitivity and protoporphyric hepatopathy 217, 218. The presumed mechanism is related to a majority of heme synthesis in the bone marrow, thus allowing for a possibility of curative treatment in some case reports, particularly as some case reports show recurrence of erythropoietic protoporphyria activity leading to fatal outcomes rapidly 219, 218, 220.
Erythropoietic protoporphyria prognosis
Erythropoietic protoporphyria (EPP) is a lifelong disease. Liver damage is the major risk, so regular liver follow-up is important. Up to 5% of affected individuals may develop more advanced liver disease, most notably cholestatic liver failure 171, 2. In most of these individuals, underlying liver cirrhosis is already present; however, some may present with rapidly progressive cholestatic liver failure 2.
Life expectancy is usually normal in patients with EPP unless liver damage develops due to hepatotoxic effects of protoporphyrins that lead to liver dysfunction. EPP generally does not decrease life expectancy but does have a great influence on the quality of life. Since the pain after photosensitivity is intense and acute, it is necessary for the patient to modify lifestyle and employment 221.
Annual assessment of red blood cell (erythrocyte) protoporphyrin levels (free and zinc-chelated), hematologic indices, and iron profile including iron, total iron binding capacity (TIBC), and ferritin is appropriate.
Liver function test including aspartate aminotransferase (AST), alanine aminotransferase (AST), total bilirubin, alkaline phosphatase (ALP) should be monitored every 6 to 12 months. Liver imaging studies including abdominal ultrasound are indicated if gallstones (cholelithiasis) is suspected.
Vitamin D 25-OH levels should be monitored in all patients whether or not they are receiving supplements.
X-Linked Protoporphyria
X-Linked Protoporphyria (XLP) also called X-linked dominant protoporphyria or X-linked dominant erythropoietic protoporphyria is an extremely rare genetic disorder of the heme-biosynthetic pathway known as the porphyrias that usually present in early life with nonblistering painful and abnormal sensitivity to the sun (photosensitivity) that can cause severe pain, burning, and itching of sun-exposed skin 222, 138, 223, 224, 225, 137. X-linked protoporphyria (XLP) symptoms may occur immediately or shortly after exposure to the sun, including direct exposure or indirect exposure such as sunlight that passes through window glass or that is reflected off water or sand. Redness and swelling of affected areas can also occur. Blistering and severe scarring occur infrequently. Chronic episodes of photosensitivity may lead to changes in the skin of sun-exposed areas. Some individuals with X-linked protoporphyria (XLP) eventually develop potentially severe liver disease.
X-linked protoporphyria (XLP) is caused by gain-of-function mutations to the 5-aminolevulinate synthase 2 (ALAS2) gene located on the X chromosome and is inherited as an X-linked dominant trait 138. Males often develop a severe form of the disorder while females may not develop any symptoms (asymptomatic) or can develop a form as severe as that seen in males. The ALAS2 gene is located on the short arm (p) of the X chromosome (Xp11.21)*. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 137. The 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme is found only in developing red blood cells found in bone marrow that develop into red blood cells (erythrocytes) called erythroblasts. 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme also plays an important role in the production of heme. Heme is a component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. The production of heme is a multi-step process that requires 8 different enzymes. ALA-synthase (ALAS2) enzyme is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA or ALA) (see Figures 5 and 6 above). Mutations of the ALAS2 gene lead to the overproduction of ALA-synthase or 5′-aminolevulinate synthase 2 enzyme, which, in turn, results in the rate of delta-aminolevulinic acid (δ-ALA or ALA) formation being increased, and the insertion of iron into protoporphyrin IX (PPIX) by ferrochelatase (FECH) the last step in heme synthesis becomes the rate-limiting for heme synthesis in erythroid tissues (the tissues where red blood cells or erythrocytes are produced and develop, in adults, the primary erythroid tissue is the bone marrow) resulting in accumulation of protoporphyrin IX (PPIX) in the the bone marrow 226, 227. If protoporphyrin IX (PPIX) compounds build up in erythroblasts (a precursor to red blood cells in the bone marrow), they can leak out and be transported through the bloodstream to your skin and other tissues such as your liver 226, 227. High levels of protoporphyrin IX (PPIX) in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP) 228, 229. For example, when protoporphyrins absorb energy from sunlight, they enter an excited state (photoactivation) and this abnormal activation results in the characteristic damage to the skin. Accumulation of protoporphyrins in the liver causes toxic damage to the liver and may contribute to the formation of gallstones. Protoporphyrin is formed within red blood cells in the bone marrow and then enters the blood plasma, which carries it to the skin where it can be photoactivated by sunlight and cause damage. The liver removes protoporphyrin from the blood plasma and secretes it into the bile.
Protoporphyrin IX (PPIX) is released from the bone marrow into the circulating red blood cells and plasma where it is taken up by the liver and vascular endothelium including the superficial skin vasculature. The protoporphyrin IX (PPIX) molecules are photodynamic (light sensitive chemical that is activated by light) and absorb light radiation in visible blue-violet light in the Soret band and to a lesser degree in the long-wave UV region 230, 124. When protoporphyrin IX (PPIX) molecules absorb light they enter an excited energy state. This energy is presumably released as fluorescence and by formation of singlet oxygen and other oxygen radicals that can produce tissue and vessel damage secondary to activation of the complement system 138. The release of histamines, kinins, and chemotactic factors may bring about skin damage 231. Accumulated liver (hepatic) protoporphyrin IX (PPIX) can precipitate in liver cells (hepatocytes) and small bile ducts (bile canaliculi) that collect bile secreted by these liver cells (hepatocytes), causing liver toxicity, decreased bile formation and flow, and cholestatic liver failure in some patients 232, 233. Large amounts of porphyrins in the gallbladder can also cause gallstones. Less commonly, a buildup of porphyrins in the liver can result in liver damage that leads to cirrhosis and liver failure.
X-linked protoporphyria (XLP) accounts for 2 to 10% of protoporphyria cases with about 2% of cases in Europe and approximately 10% of cases in the United States 119, 179, 234.
The diagnosis of X-linked protoporphyria (XLP) is established in a male index case (the affected individual through whom a family with a genetic disorder is ascertained) with markedly increased free red blood cell (erythrocyte) protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) by identification of a hemizygous pathogenic gain-of-function variant in ALAS2 on molecular genetic testing.
The diagnosis of X-linked protoporphyria (XLP) is established in a female index case with increased free erythrocyte protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) by identification of a heterozygous pathogenic gain-of-function variant in ALAS2 on molecular genetic testing.
The treatment of X-linked protoporphyria is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with X-linked protoporphyria (XLP). Because the disorder is so rare, most treatment information is based on erythropoietic protoporphyria (EPP), which is clinically similar to X-linked protoporphyria (XLP).
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, unless they contain light-reflective ingredients (e.g., zinc oxide). Some tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Avoidance of sunlight can potentially cause vitamin D deficiency and some individuals may require supplemental vitamin D.
A high potency form of oral beta-carotene (Lumitene) may be given to improve an affected individual’s tolerance of sunlight. This drug causes skin discoloration and may improve tolerance to sunlight. Oral Lumitene (beta-carotene) (120–180 mg/dL) has been used to improve tolerance to sunlight if the dose is adjusted to maintain serum carotene levels in the range of 10-15 μmol/L (600–800 μg/dL), causing mild skin discoloration due to carotenemia. The beneficial effects of beta-carotene (Lumitene) may involve quenching of singlet oxygen or free radicals. However, a systematic review of about 25 studies showed that the available data are unable to prove efficacy of treatments including beta-carotene, N-acetyl cysteine, and vitamin C 210. For more information on oral beta-carotene (Lumitene) treatment, contact the American Porphyria Foundation and the Prophyria Consortium of the Rare Diseases Clinical Research Network.
Another drug sometimes used to improve tolerance to sunlight is cysteine.
In 2019, the Food and Drug Administration (FDA) approved Afamelanotide (Scenesse®) for the treatment of adult patients with erythropoietic protoporphyria (EPP). Afamelanotide (Scenesse) is a controlled-release, long-acting injectable implant, alpha-melanocyte-stimulating hormone (α-MSH) analogue, that increases eumelanin by binding to the melanocortin-1 receptor and provides sun protection and improves sun tolerance by increasing skin pigmentation and antioxidant properties 235, 236. Afamelanotide (Scenesse) was available in Europe for a period of time before its approval in the United States. Afamelanotide (Scenesse) was approved for patients with the erythropoietic protoporphyria (EPP) by the European Medicines Agency in 2014, and by the FDA in October 2019. Afamelanotide showed positive results in Phase III clinical trials in the US and Europe 237. Long-term studies in Europe show good compliance, clinical effectiveness, and improved quality of life 238.
Other treatment is symptomatic and supportive. Individuals with high levels of protoporphyrin in the plasma and red blood cells should be observed closely by a physician for possible liver malfunction that could eventually lead to liver failure.
When iron deficiency is present, iron supplements may be given. A drug called Prevalite (cholestyramine) or activated charcoal maybe prescribed to interrupt the circulation of protoporphyrin through the liver and intestines in patients with liver disease.
Liver transplantation has been performed as a life-saving measure in patients with erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) related liver failure. Bone marrow transplant can also be performed after liver transplant to prevent further damage to the liver.
X-linked protoporphyria (XLP) patients should also receive vaccination against hepatitis A and B to prevent other causes of liver damage.
X-linked protoporphyria (XLP) patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
X-linked protoporphyria causes
X-linked protoporphyria (XLP) is caused by gain-of-function mutations to the 5-aminolevulinate synthase 2 (ALAS2) gene located on the X chromosome and is inherited as an X-linked dominant trait 138. Males often develop a severe form of the disorder while females may not develop any symptoms (asymptomatic) or can develop a form as severe as that seen in males. The ALAS2 gene is located on the short arm (p) of the X chromosome (Xp11.21)*. The ALAS2 gene provides instructions for making an enzyme called 5′-aminolevulinate synthase 2 or erythroid ALA-synthase 137. The 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme is found only in developing red blood cells found in bone marrow that develop into red blood cells (erythrocytes) called erythroblasts. 5′-aminolevulinate synthase 2 or erythroid ALA-synthase (ALAS2) enzyme also plays an important role in the production of heme. Heme is a component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. The production of heme is a multi-step process that requires 8 different enzymes. ALA-synthase (ALAS2) enzyme is responsible for the first step in this process, the formation of a compound called delta-aminolevulinic acid (δ-ALA or ALA). Mutations of the ALAS2 gene lead to the overproduction of ALA-synthase or 5′-aminolevulinate synthase 2 enzyme, which, in turn, results in the rate of delta-aminolevulinic acid (δ-ALA or ALA) formation being increased, and the insertion of iron into protoporphyrin IX (PPIX) by ferrochelatase (FECH) the last step in heme synthesis becomes the rate-limiting for heme synthesis in erythroid tissues (the tissues where red blood cells or erythrocytes are produced and develop, in adults, the primary erythroid tissue is the bone marrow) resulting in accumulation of protoporphyrin IX (PPIX) in the the bone marrow 226, 227. If protoporphyrin IX (PPIX) compounds build up in erythroblasts (a precursor to red blood cells in the bone marrow), they can leak out and be transported through the bloodstream to your skin and other tissues such as your liver 226, 227. High levels of protoporphyrin IX (PPIX) in the skin cause the oversensitivity to sunlight that is characteristic of X-linked protoporphyria (XLP) 228, 229. For example, when protoporphyrins absorb energy from sunlight, they enter an excited state (photoactivation) and this abnormal activation results in the characteristic damage to the skin. Accumulation of protoporphyrins in the liver causes toxic damage to the liver and may contribute to the formation of gallstones. Protoporphyrin is formed within red blood cells in the bone marrow and then enters the blood plasma, which carries it to the skin where it can be photoactivated by sunlight and cause damage. The liver removes protoporphyrin from the blood plasma and secretes it into the bile.
Chromosomes are found in the nucleus of all body cells. They carry the genetic characteristics of each individual. Pairs of human chromosomes are numbered from 1 through 22, with an unequal 23rd pair of X and Y chromosomes for males and two X chromosomes for females. Each chromosome has a short arm designated as “p” and a long arm identified by the letter “q”. Chromosomes are further subdivided into bands that are numbered. For example, “chromosome Xp22.2-22.1” refers to bands 22.2 through 22.1 on the short arm of chromosome X.
X-linked protoporphyria (XLP) results from gain-of-function mutations in erythroid-specific ALAS2 136. Mutations associated with X-linked protoporphyria (XLP) have only been observed in exon 11, which encodes the C-terminus, and result in a gain-of-function of ALAS2. These mutations result in stop or frameshift lesions that prematurely truncate or abnormally elongate the wild-type enzyme, leading to increased ALAS2 activity 136, 234. In X-linked protoporphyria (XLP), all males are affected 222. In heterozygous females (females with one normal copy and one mutated copy of ALAS2 gene on their X chromosomes) with X-linked protoporphyria (XLP), the random X-inactivation pattern directly influences the penetrance and the severity of the phenotype. X-linked protoporphyria (XLP) females can be asymptomatic clinically with normal protoporphyrins, be asymptomatic clinically with slightly elevated protoporphyrin levels or have significant symptoms based on the pattern of X-inactivation 239, 222.
X-linked protoporphyria inheritance pattern
X-linked protoporphyria (XLP) is passed down through families in an X-linked manner 127. Males have one X chromosome and one Y chromosome, while females have two X chromosomes.This means that males have only one copy of the ALAS2 gene and females have two copies of the ALAS2 gene. When a male has a mutation is his single copy of ALAS2 gene, he is expected to have symptoms of X-linked protoporphyria (XLP). In a woman with a mutation in one of her ALAS2 genes, the second functioning copy of the ALAS2 gene can help compensate and may lead to less severe symptoms or no symptoms at all. It is not possible to predict or control the severity of X-linked protoporphyria (XLP) in females. Men with XLP pass on their X chromosome to their daughters and their Y chromosome to their sons. Therefore, a man with X-linked protoporphyria (XLP) with pass on his genetic change to all his daughters, and none of his sons. For a female with XLP, she will pass on the X chromosome with the genetic change 50% of the time. Thus, in each pregnancy, there is a 50% chance of having a child with a mutation in ALAS2 gene.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 20. X-linked protoporphyria (XLP) inheritance pattern
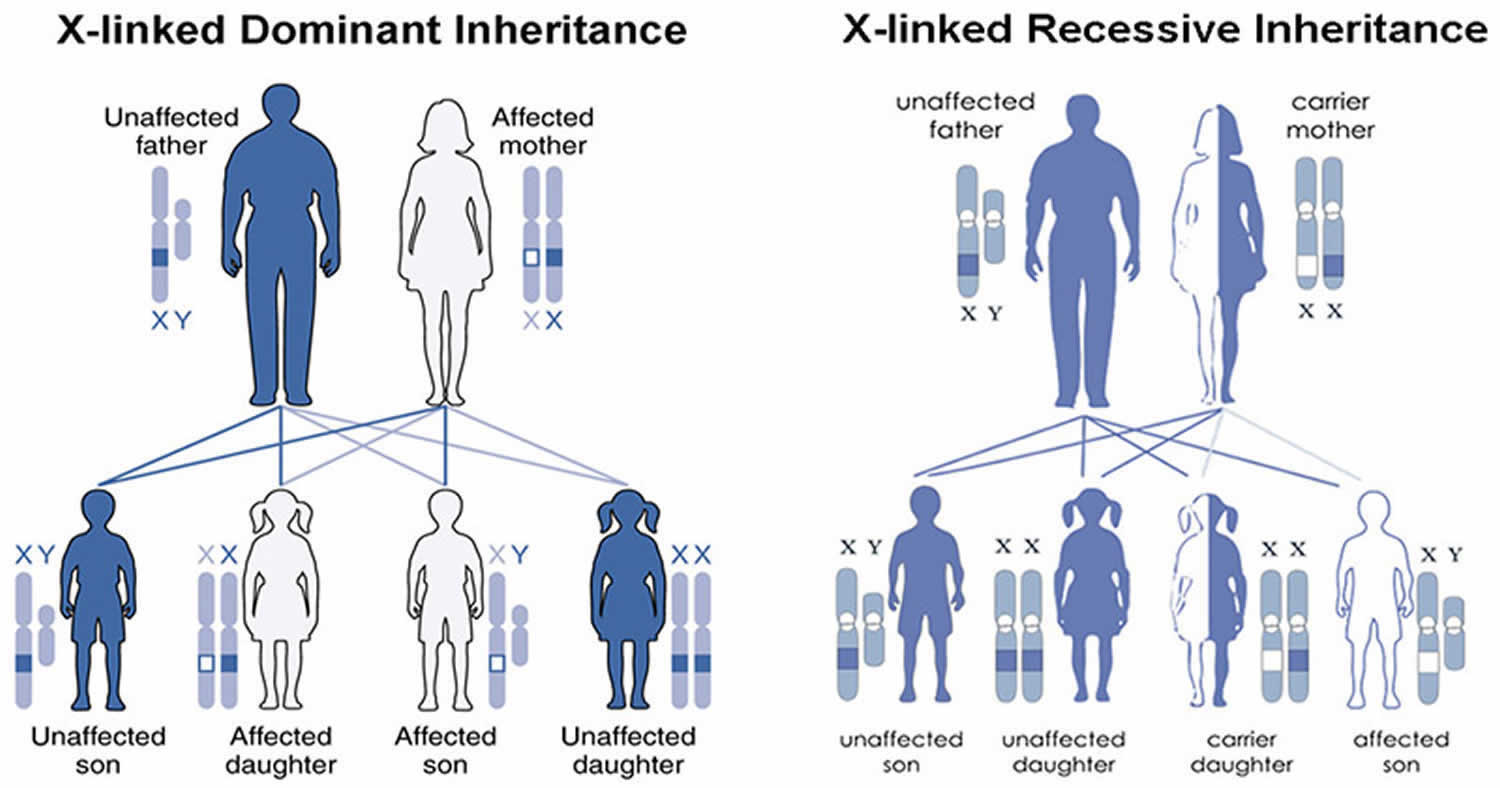
X-Linked Protoporphyria Pathophysiology
Bone marrow reticulocytes are thought to be the primary source of the accumulated protoporphyrin IX (PPIX) that is excreted in bile and feces 222. Most of the excess protoporphyrin IX (PPIX) in circulating erythrocytes is found in a small percentage of cells, and the rate of protoporphyrin IX (PPIX) leakage from these cells is proportional to their protoporphyrin content.
The skin of persons with X-linked protoporphyria (XLP) is maximally sensitive to visible blue-violet light near 400 nm, which corresponds to the so-called “Soret band” (the narrow peak absorption maximum that is characteristic for protoporphyrin and other porphyrins) 222. When porphyrins absorb light they enter an excited energy state. This energy is presumably released as fluorescence and by formation of singlet oxygen and other oxygen radicals that can produce tissue and vessel damage. This may involve lipid peroxidation, oxidation of amino acids, and cross-linking of proteins in cell membranes 222.
Photoactivation of the complement system and release of histamine, kinins, and chemotactic factors may mediate skin damage 222. Histologic changes occur predominantly in the upper dermis and include deposition of amorphous material containing immunoglobulin, complement components, glycoproteins, glycosaminoglycans, and lipids around blood vessels. Damage to capillary endothelial cells in the upper dermis has been demonstrated immediately after light exposure in this disease 240.
Long-term observations of individuals with protoporphyria generally show little change in protoporphyrin levels in erythrocytes, plasma, and feces 241. In contrast, severe liver complications, when they occur, often follow increasing accumulation of protoporphyrin in erythrocytes, plasma, and liver. Iron deficiency and factors that impair liver function sometimes contribute. Enterohepatic circulation of protoporphyrin may favor its return and retention in the liver, especially when liver function is impaired. Liver damage probably results at least in part from protoporphyrin accumulation itself 222. As this porphyrin is insoluble, it tends to form crystalline structures in liver cells, can impair mitochondrial functions in liver cells, and can decrease hepatic bile formation and flow 242.
X-linked protoporphyria signs and symptoms
Hypersensitivity of the skin to sunlight beginning in infancy or childhood is the characteristic finding of X-linked protoporphyria (XLP) 228, 222. Most patients develop symptoms within 30 minutes of sun exposure. Affected individuals develop tingling, burning, pain, and itching of the skin after exposure to sunlight. Sometimes these symptoms are accompanied by swelling and redness (erythema) of the affected areas 228, 222. Large blisters and severe scarring, which are common to other forms of cutaneous porphyria, usually do not occur in individuals with X-linked protoporphyria 222. The back of the hands and face are most commonly affected but any sun exposed area can be affected. Symptoms may be noticed as quickly as a few minutes after exposure to the sun 243. Although most symptoms usually subside within 24-48 hours (may take up to 4–7 days), pain and a red or purple discoloration of the skin may persist for several days after the initial incident 243, 180. Pain is disproportionately severe in relation to the visible skin lesions. Pain associated with X-linked protoporphyria can be excruciating and is often resistant to pain medications, even narcotics.
Photosensitivity is lifelong. Repeated episodes of photosensitivity may eventually causes changes in the skin of affected individuals. Such changes include thickening and hardening of the sun-exposed skin (lichenification), development of a rough or leathery texture, small facial pock-like pits, and grooving around the lips and loss of lunulae of the nails 231, 222.
Some individuals with X-linked protoporphyria develop liver disease, which can range from mild liver abnormalities to liver failure 222. Information on liver disease is limited, but the risk of liver disease is believed to be higher in X-linked protoporphyria than in erythropoietic protoporphyria (EPP). Affected individuals may experience back pain and severe abdominal pain especially in the upper right area of the abdomen. In some affected individuals, the flow of bile through the gallbladder and bile ducts may be interrupted (cholestasis) leading to gallstones. These stones can cause obstruction and inflammation of the gallbladder (cholecystitis). Scarring of the liver (cirrhosis) may also develop and some individuals may eventually develop end stage liver failure.
Additional symptoms have been reported in individuals with X-linked protoporphyria including mild anemia (low levels of circulating red blood cells) and iron deficiency.
The signs and symptoms in heterozygous females (females with one normal copy and one mutated copy of ALAS2 gene on their X chromosomes) ranges from asymptomatic to as severe as in affected males 222.
Figure 21. X-linked protoporphyria
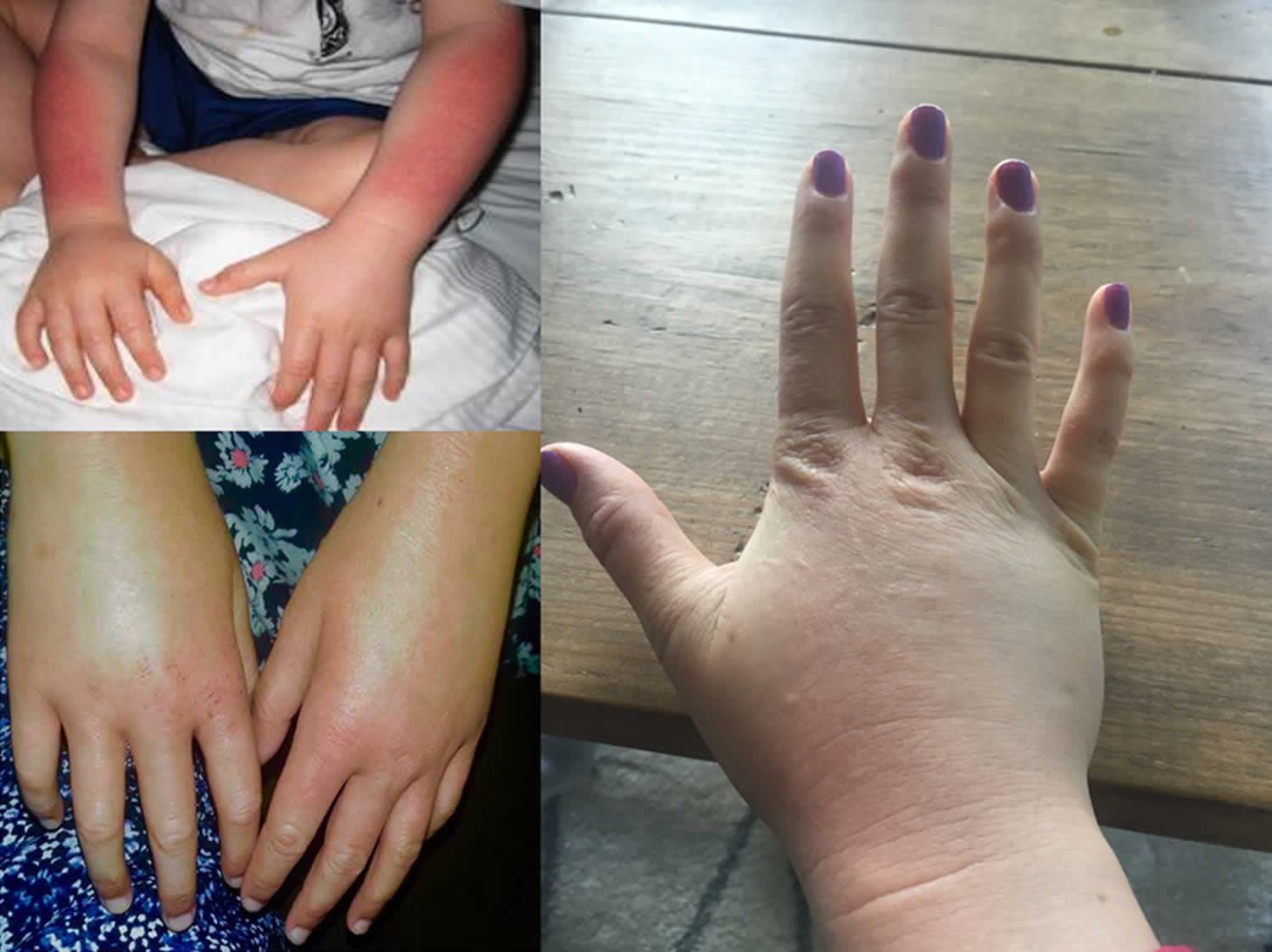
Footnote: Skin redness (erythema) and swelling (edema) on the back of hands and forearm seen after acute phototoxic episode.
[Source 138 ]Liver disease
The excess protoporphyrin in erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) is excreted by the liver into the bile where it enters the enterohepatic circulation 170. Progressive accumulation of protoporphyrin IX (PPIX) may occur in the liver when the biliary excretion does not keep pace with the load being presented to the liver. When hepatocellular damage reaches a critical stage, protoporphyrin IX (PPIX) accumulation will rapidly accelerate due to marked impairment of biliary excretion 233, 2, 222. Concomitant conditions such as viral hepatitis, excessive alcohol consumption, and use of drugs which induce cholestasis may contribute to worsening liver disease. End-stage liver disease is typically preceded by an elevation in plasma and erythrocyte protoporphyrin IX (PPIX) levels. Patients may also develop a motor neuropathy in the setting of liver failure 213, 244.
The excess amounts of free protoporphyrin IX (PPIX) may become insoluble and aggregate in the hepatocytes and small biliary canaliculi leading to obstruction to bile flow and cholestasis. About 20–30% of patients with erythropoietic protoporphyria (EPP) will have elevations in serum aminotransferases20. Protoporphyrin in bile may also crystallize forming gallstones. In one series, cholelithiasis were seen in 23.5% of patients 180.
Anemia
Mild anemia, typically microcytic anemia can be seen in erythropoietic protoporphyria (EPP) patients 146. Patients with erythropoietic protoporphyria (EPP) appear to have an abnormal iron metabolism but the mechanism of iron deficiency is unclear 245, 201. The cause of microcytic anemia and low iron and ferritin levels in erythropoietic protoporphyria (EPP) patients is unknown 200, 246. Previous studies suggest that erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) patients have normal iron absorption and an appropriate hepcidin response 247. The iron deficiency in erythropoietic protoporphyria (EPP) and X-linked protoporphyria (XLP) patients does not appear to be related to chronic inflammation or iron loss. The cause and mechanism of iron deficiency in these patients remains to be resolved.
Vitamin D deficiency
X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP) patients can develop vitamin D deficiency secondary to sun avoidance 248, 249. A recent report showed that the prevalence of osteopenia (low bone mineral density) and osteoporosis is increased in patients in erythropoietic protoporphyria (EPP) 250.
X-linked protoporphyria diagnosis
A diagnosis of X-linked protoporphyria is based upon identification of characteristic symptoms (e.g., non-blistering photosensitivity), a detailed patient history, a thorough clinical evaluation, and a variety of specialized tests 228, 251, 252, 222.
X-linked protoporphyria (XLP) should be suspected in individuals with the following clinical findings and initial laboratory findings 222:
- Clinical findings
- Skin photosensitivity, usually beginning in childhood
- Burning, tingling, pain, and itching of the skin (the most common findings); may occur within minutes of sun/light exposure, followed later by erythema and swelling
- Painful symptoms; may occur without obvious skin damage
- Absent or sparse blisters and bullae
- Note: The absence of skin damage (e.g., scarring), vesicles, and bullae often make it difficult to suspect the diagnosis.
- Liver complications, particularly cholestatic liver disease, may develop in fewer than 5% of affected individuals.
A diagnosis of X-linked protoporphyria may be made through blood tests that can detect markedly increased levels of metal-free and zinc-bound protoporphyrins within red blood cells (erythrocytes). A higher ratio of zinc-bound protoporphyrin to metal-free protoporphyrin can differentiate X-linked protoporphyria from erythropoietic protoporphyria.
Note: It is essential to use an assay for erythrocyte protoporphyrin that distinguishes between free protoporphyrin and zinc-chelated protoporphyrin to differentiate X-linked protoporphyria (XLP) from erythropoietic protoporphyria (EPP) and several other conditions that may lead to elevation of erythrocyte protoporphyrins.
- Initial laboratory findings. Detection of markedly increased free erythrocyte protoporphyrin IX (PPIX) and zinc-chelated erythrocyte protoporphyrin IX (PPIX) is the most sensitive biochemical diagnostic test for X-linked protoporphyria (XLP).
- Erythroid-specific 5-aminolevulinate synthase 2 (ALAS2) enzyme activity >100% of normal
- Free protoporphyrin/zinc-chelated protoporphyrin ratio 90:10 to 50:50
- Urine Protoporphyrins not detectable
- Stool Protoporphyrin normal or increased
- Plasma porphyrins increased
Molecular genetic testing can confirm a diagnosis of X-linked protoporphyria (XLP) by detecting mutations in the ALAS2 gene (the only gene known to cause this disorder).
Additional tests may be performed such as blood tests to evaluate anemia and iron stores in the body and vitamin D levels, or an abdominal sonogram to detect and evaluate liver disease potentially associated with X-linked protoporphyria.
Newer imaging modalities such as Fibroscan® may be useful in evaluating liver fibrosis; however, this has not been validated in erythropoietic protoporphyria (EPP) or X-linked protoporphyria (XLP) 222.
X-linked protoporphyria treatment
The treatment of X-linked protoporphyria is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with X-linked protoporphyria (XLP). Because the disorder is so rare, most treatment information is based on erythropoietic protoporphyria (EPP), which is clinically similar to X-linked protoporphyria (XLP).
Avoidance of sunlight will benefit affected individuals and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, unless they contain light-reflective ingredients (e.g., zinc oxide). Some tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Avoidance of sunlight can potentially cause vitamin D deficiency and some individuals may require supplemental vitamin D.
A high potency form of oral beta-carotene (Lumitene) may be given to improve an affected individual’s tolerance of sunlight. This drug causes skin discoloration and may improve tolerance to sunlight. Oral Lumitene (beta-carotene) (120–180 mg/dL) has been used to improve tolerance to sunlight if the dose is adjusted to maintain serum carotene levels in the range of 10-15 μmol/L (600–800 μg/dL), causing mild skin discoloration due to carotenemia. The beneficial effects of beta-carotene (Lumitene) may involve quenching of singlet oxygen or free radicals. However, a systematic review of about 25 studies showed that the available data are unable to prove efficacy of treatments including beta-carotene, N-acetyl cysteine, and vitamin C 210. For more information on oral beta-carotene (Lumitene) treatment, contact the American Porphyria Foundation and the Prophyria Consortium of the Rare Diseases Clinical Research Network.
Another drug sometimes used to improve tolerance to sunlight is cysteine.
In 2019, the Food and Drug Administration (FDA) approved Afamelanotide (Scenesse®) for the treatment of adult patients with erythropoietic protoporphyria (EPP). Afamelanotide (Scenesse) is a controlled-release, long-acting injectable implant, alpha-melanocyte-stimulating hormone (α-MSH) analogue, that increases eumelanin by binding to the melanocortin-1 receptor and provides sun protection and improves sun tolerance by increasing skin pigmentation and antioxidant properties 235, 236. Afamelanotide (Scenesse) was available in Europe for a period of time before its approval in the United States. Afamelanotide (Scenesse) was approved for patients with the erythropoietic protoporphyria (EPP) by the European Medicines Agency in 2014, and by the FDA in October 2019. Afamelanotide showed positive results in Phase III clinical trials in the US and Europe 237. Long-term studies in Europe show good compliance, clinical effectiveness, and improved quality of life 238.
Other treatment is symptomatic and supportive.
X-linked protoporphyria (XLP) patients should be seen at least yearly to monitor protoporphyrin levels, anemia, liver enzymes, iron and vitamin D levels.
Liver disease treatment
Treatment of liver disease and complications, which may be accompanied by motor neuropathy, is difficult.
- Cholestyramine and other porphyrin absorbents, such as activated charcoal, may interrupt the enterohepatic circulation of protoporphyrin and promote its fecal excretion, leading to some improvement 212.
- Cholestyramine absorbs porphyrin. Cholestyramine may interrupt the recirculation of protoporphyrin secreted into the bile back into the liver and promote its excretion through the feces. Other drugs that absorb porphyrins such as activated charcoal have also been used to treat affected individuals.
- Cholestyramine and other porphyrin absorbents, such as activated charcoal may lead to improvement of liver disease.
- Plasmapheresis and intravenous hemin have been used to treat people with erythropoietic protoporphyria (EPP) 211.
- Liver transplantation has been performed as a lifesaving measure in individuals with severe protoporphyric liver disease 213, 214. However, many liver transplant recipients experience a recurrence of the protoporphyric liver disease in the transplanted liver. Combined bone marrow and liver transplantation is indicated in patients with liver failure to prevent future damage to the allografts 216, and sequential liver and bone marrow transplantation has been successful in curing protoporphyric liver disease 253.
- Bone marrow transplantation has also been attempted without liver transplantation in some instances. A child age two years with X-linked protoporphyria (XLP) and stage 4 liver fibrosis was treated with a hematopoietic progenitor cell transplantation that stabilized his liver disease, thus avoiding liver transplantation 254.
Individuals with any form of protoporphyria should avoid substances associated with cholestasis including alcohol and certain drugs such as estrogens.
In patients with cholestatic liver failure, use of protective filters for artificial lights in the operating room to prevent phototoxic damage during procedures such as endoscopy and surgery 255.
Additional treatment
- Vitamin D supplementation is advised as patients are predisposed to vitamin D deficiency resulting from sun avoidance.
- Immunizations for hepatitis A and B are recommended as well.
- Iron supplementation may be attempted in persons with X-linked protoporphyria (XLP) who have anemia and low ferritin levels. Iron supplementation therapy requires strict monitoring by physicians. Whatley et al 226 reported some evidence of diminished iron stores in males with X-linked protoporphyria (XLP); in one patient with iron deficiency, iron repletion decreased protoporphyrin accumulation and corrected the anemia. Subsequent reports indicate that iron supplementation can improve protoporphyrin levels, liver damage, and anemia in X-linked protoporphyria (XLP) 256. A pilot study using oral iron supplementation in persons with X-linked protoporphyria (XLP) showed a reduction in protoporphyrin levels, but also carries a risk of increased photosensitivity 257.
Pregnancy Management
There is no information on pregnancy management in X-linked protoporphyria (XLP). Based on experience with erythropoietic protoporphyria (EPP), pregnancy is unlikely to be complicated by X-linked protoporphyria (XLP) 258.
Investigational Therapies
A Phase 2 clinical trial with MT-7117, an oral small molecule that works as a melanocortin 1 receptor agonist and increases skin pigmentation in subjects with erythropoietic protoporphyria (EPP) has been completed 259. A Phase 3 clinical trial for adults and children is planned for MT-7117.
X-Linked Protoporphyria prognosis
The natural history of X-linked protoporphyria (XLP) is not as well characterized as that of the autosomal recessive type of erythropoietic protoporphyria (EPP) 222. A natural history study from the US described 22 individuals with X-linked protoporphyria (XLP) from seven unrelated families 180.
X-linked protoporphyria (XLP) in Males
While the skin signs and symptoms in males with X-linked protoporphyria (XLP) are similar to those of erythropoietic erythropoietic protoporphyria (EPP), Balwani et al 180 suggest that males with X-linked protoporphyria (XLP) have significantly higher protoporphyrin levels and increased risk of liver dysfunction.
Photosensitivity
Onset of photosensitivity is typically in infancy or childhood with the first exposure to sun; in most individuals with X-linked protoporphyria (XLP) the photosensitivity is lifelong.
Most males with X-linked protoporphyria (XLP) develop acute cutaneous photosensitivity within five to 30 minutes following exposure to sun or ultraviolet light. Photosensitivity symptoms are provoked mainly by visible blue-violet light in the Soret band, to a lesser degree in the long-wave UV region.
The initial symptoms reported are tingling, burning, and/or itching that may be accompanied by swelling and redness. Symptoms vary based on the intensity and duration of sun exposure; pain may be severe and refractory to narcotic analgesics, persisting for hours or days after the initial phototoxic reaction. Symptoms may seem out of proportion to the visible skin lesions. Blistering lesions are uncommon.
Affected males are also sensitive to sunlight that passes through window glass, which does not block long-wave UVA or visible light.
Skin signs and symptoms
Multiple episodes of acute photosensitivity may lead to chronic changes of sun-exposed skin (lichenification, leathery pseudovesicles, grooving around the lips) and loss of lunulae of the nails. The back of the hands is most notably affected.
Severe scarring is rare, as are hyper- or hypopigmentation, skin friability, and hirsutism.
Unlike in other cutaneous porphyrias, blistering and scarring rarely occur.
Liver problems
Protoporphyrin is not excreted in the urine by the kidneys, but is taken up by the liver and excreted in the bile. Accumulated protoporphyrin in the bile can form stones, reduce bile flow, and damage the liver. Protoporphyric liver disease may cause back pain and severe abdominal pain (especially in the right upper quadrant).
The information on X-linked protoporphyria (XLP) and liver disease is limited 222. The risk for liver dysfunction in X-linked protoporphyria (XLP) (observed in 5/31 affected individuals) is higher than the risk in erythropoietic protoporphyria (EPP) 226. A natural history study in the US showed that 40% of males with X-linked protoporphyria (XLP) had a history of abnormal liver enzymes compared to 33% of persons with erythropoietic protoporphyria (EPP). Gallstones were seen in 40% of males with X-linked protoporphyria (XLP) and 33.3% of females with X-linked protoporphyria (XLP) compared to 22.1% of individuals with erythropoietic protoporphyria (EPP).
Note that the information on liver involvement presented below is based on experience with liver disease in autosomal recessive erythropoietic protoporphyria (EPP). Gallstones composed in part of protoporphyrin may be symptomatic in individuals with X-linked protoporphyria (XLP) and need to be excluded as a cause of biliary obstruction in persons with hepatic decompensation.
Life-threatening liver complications are preceded by increased levels of plasma and erythrocyte protoporphyrins, worsening liver function tests, increased photosensitivity, and increased deposition of protoporphyrins in liver cells and bile canaliculi. End-stage liver disease may be accompanied by motor neuropathy, similar to that seen in acute porphyrias. Comorbid conditions, such as viral hepatitis, alcohol abuse, and use of oral contraceptives, which may impair hepatic function or protoporphyrin metabolism, may contribute to hepatic disease in some 213.
Blood problems
Anemia and abnormal iron metabolism can occur in X-linked protoporphyria (XLP). Mild anemia with microcytosis and hypochromia or occasionally reticulocytosis can be seen 222. However, hemolysis is absent or mild 222. In a recent series, 30% of males with X-linked protoporphyria (XLP) and 75% of females with X-linked protoporphyria (XLP) were anemic 180.
Vitamin D deficiency
Individuals with X-linked protoporphyria (XLP) who avoid sun/light are at risk for vitamin D deficiency 248, 249, 146.
Triggering factors
Unlike the triggering factors for acute hepatic porphyrias, the only known precipitating factor for X-linked protoporphyria (XLP) is sunlight.
X-linked protoporphyria (XLP) in Females
The signs and symptoms of X-linked protoporphyria (XLP) in heterozygous females, the consequence of random X-chromosome inactivation, ranges from as severe as in affected males to asymptomatic. The median age of symptom onset for females with X-linked protoporphyria (XLP) was 11 years. Following sun exposure, symptom onset ranged from within ten minutes to none 180.
X-Linked Protoporphyria life expectancy
People with X-linked protoporphyria (XLP) generally have a life expectancy similar to those without porphyria, except for those with advanced liver disease 222.
Congenital erythropoietic porphyria
Congenital erythropoietic porphyria (CEP) also called Gunther’s disease is an extremely rare inherited autosomal recessive disorder that involve defects in heme or ‘haem’ biosynthetic pathway caused by mutations (changes) in the UROS gene which results in low levels of the enzyme uroporphyrinogen III cosynthase or uroporphyrinogen-III synthase (UROS), the fourth enzyme in the heme biosynthetic pathway 260, 261, 262, 263, 264, 265, 266, 267, 268, 269, 270, 271, 272, 273, 274, 275, 276, 277, 278, 279. The UROS (uroporphyrinogen III synthase or uroporphyrinogen III cosynthase) enzyme is involved in the production of a molecule called heme (haem). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). The production of heme is a multi-step process that requires 8 different enzymes. In the presence of insufficient uroporphyrinogen III synthase (UROS) activity, hydroxymethylbilane (HMB), the product of the third step spontaneously closes and forms the non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 280. Coproporphyrinogen I then oxidizes spontaneously to form coproporphyrin I and uroporphyrin I, two harmful and non-physiological compounds associated with abnormalities involving various organs 260. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors (erythroblasts) in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Your bone marrow, the spongy tissue inside your bones, is essential for producing blood cells and supporting the immune system. Your bone marrow acts as a factory for red blood cells, white blood cells, and platelets, which are vital for oxygen transport, fighting infection, and blood clotting, respectively. Your bone marrow also plays a role in storing fat and supporting bone and cartilage health. As these toxic porphyrins accumulate in your bone marrow, they may be released into your blood and subsequently found in the plasma and mature red blood cells. They then may travel to your skin, and upon exposure to light, generate free radicals that can damage cells and tissues. This leads to disease characterized by severe skin photosensitivity and breakdown of red blood cells (hemolysis) 281. The major symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of your skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in your skin cause skin friability and blistering (bullae) and the fluid-filled sacs may rupture and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and in severe instances, this can lead to skin mutilation and deformities 282. Your face, ears, neck, arms, forearms and hands are the most commonly affected areas.
In most cases, congenital erythropoietic porphyria (CEP) is caused by UROS gene mutations located on chromosome 10q26.2 and follows an autosomal recessive inheritance pattern 283, 284. Typically, there is no family history of the disease. Neither parent has symptoms of congenital erythropoietic porphyria (CEP), but each carries a defective UROS gene that they can pass to their children. Affected offspring have two copies of the defective UROS gene, one inherited from each parent.
Rarely congenital erythropoietic porphyria (CEP) is caused by GATA1 gene mutation located on the X chromosome (chromosome Xp11.23) that affect males 285, 274, 286. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 274, 286.
In a small percentage of congenital erythropoietic porphyria patients (>10%), a disease-causing mutation has not been detected in the UROS or GATA1 genes, raising the possibility that additional gene(s) may play a role in congenital erythropoietic porphyria pathogenesis 287.
Disease-causing mutations in either UROS gene or GATA1 gene result in absent or markedly reduced UROS (uroporphyrinogen III synthase) enzymatic activity. UROS (uroporphyrinogen III synthase) enzyme catalyzes the cyclization of hydroxymethylbilane (HMB) with concomitant inversion of ring D to yield uroporphyrinogen III (URO III) 288. Deficiency of UROS (uroporphyrinogen III synthase) enzyme results in accumulation of HMB (hydroxymethylbilane), most of which is converted nonenzymatically to non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 280. Coproporphyrinogen I then oxidizes spontaneously to form the porphyrins coproporphyrin I and uroporphyrin I. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Since these toxic porphyrins are photocatalytic compounds, exposure of the skin to sunlight and other sources of long-wave ultraviolet light elicits a phototoxic reaction, resulting in blistering and vesicle formation as well as increased friability of the skin and breakdown of red blood cells (hemolysis) 281, 289, 290.
Congenital erythropoietic porphyria (CEP) is a very rare genetic disorder affecting less than 1 in 1,000,000 children and it affects males and females in equal numbers. So far, about 280 congenital erythropoietic porphyria patients have been described in the literature, primarily in Western countries 287, 291, 292 and their clinical manifestations are markedly heterogeneous, ranging from non-immune hydrops fetalis to milder, later-onset forms characterized by mild cutaneous involvement without hematologic symptoms in adult life 290, 293. Severely affected patients are transfusion-dependent throughout life, have secondary hypersplenism and significant cutaneous involvement. Severe complications such as secondary skin infections with subsequent bone resorption and photomutilation, leading to loss of digits and facial features are common 281. Chronic infections have caused osteomyelitis in some congenital erythropoietic porphyria (CEP) patients 294, 295.
Congenital erythropoietic porphyria (CEP) onset in most affected individuals occurs at birth or early infancy. The first sign of congenital erythropoietic porphyria (CEP) is often pink-to-dark red discoloration of the urine. Hemolytic anemia is common and can range from mild to severe, with some affected individuals requiring chronic blood transfusions 260. Porphyrin deposition may lead to corneal ulcers and scarring, reddish-brown discoloration of the teeth (erythrodontia), and bone loss and/or expansion of the bone marrow. However, congenital erythropoietic porphyria (CEP) signs and symptoms are broad and range from nonimmune hydrops fetalis in utero to late-onset disease with only mild skin manifestations in adulthood.
The extent of enzyme deficiency caused by the UROS gene mutation in each case is the major determinant of the age of onset and severity of symptoms seen in the disease 260.
The diagnosis of congenital erythropoietic porphyria may be suspected when the reddish-colored urine is noted at birth or later in life. This finding, or the occurrence of skin blisters on sun or light exposure, should lead to a thorough clinical evaluation and specialized laboratory tests. The diagnosis can be made by testing the urine for increased levels of specific porphyrins. Diagnostic confirmation requires the demonstration of the specific UROS enzyme activity and/or by identifying the specific mutation(s) in the UROS gene which is/are responsible for the impaired enzyme.
Prenatal and preimplantation genetic diagnoses are available for subsequent pregnancies in congenital erythropoietic porphyria families.
Figure 22. Congenital erythropoietic porphyria (CEP) signs and symptoms
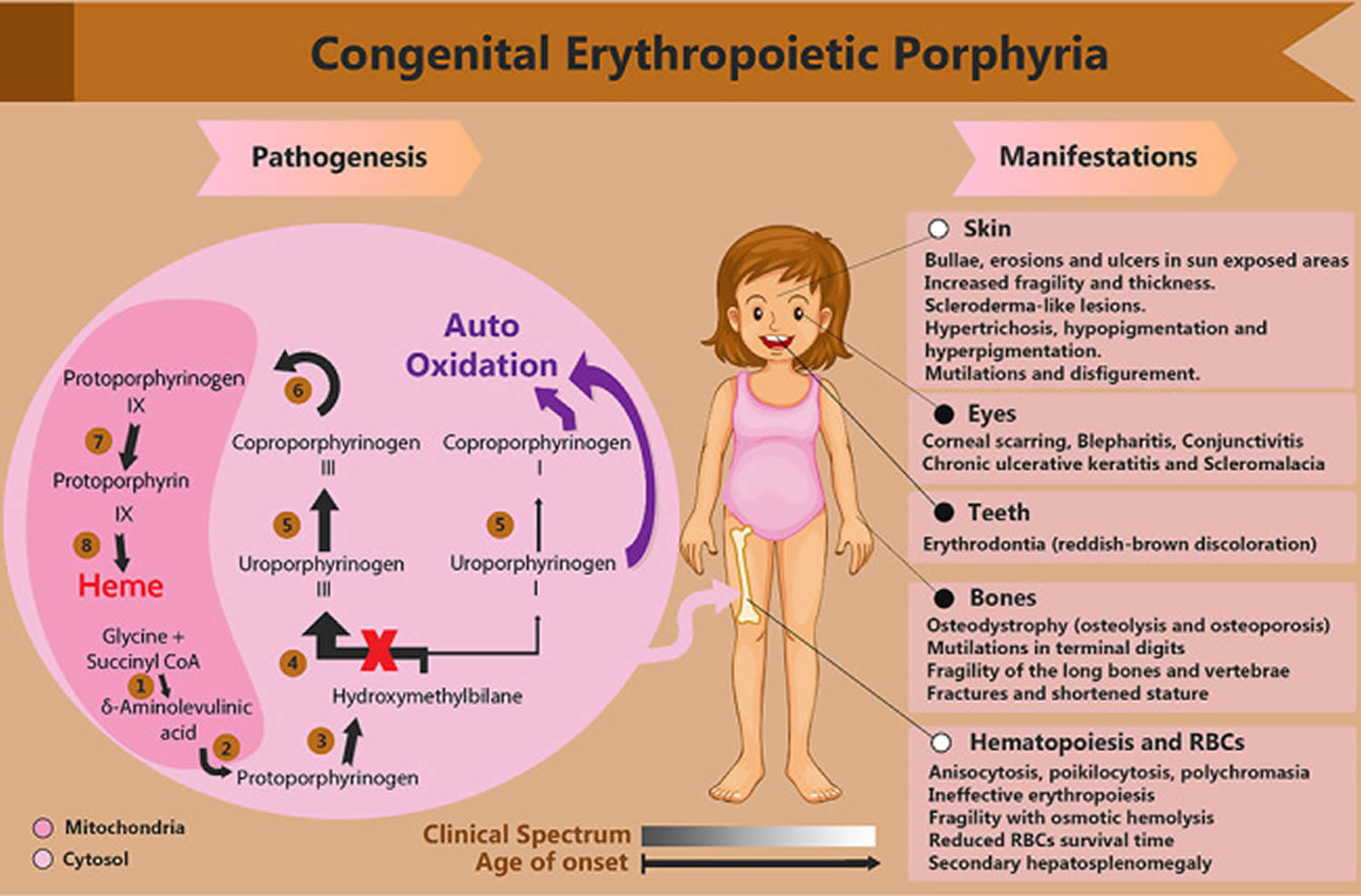
Footnotes: An illustration of the heme biosynthesis pathway and congenital erythropoietic porphyria (CEP) pathophysiology. Each number represents the enzyme of the corresponding step: (1) aminolevulinic acid synthase (ALAS), (2) aminolevulinic acid dehydratase (ALAD), (3) hydroxymethylbilane synthase (HMBS), (4) uroporphyrinogen III synthase (UROS), (5) uroporphyrinogen decarboxylase (UROD), (6) coproporphyrinogen-III oxidase (CPOX), (7) protoporphyrinogen oxidase (PPOX), (8) ferrochelatase (FECH). The gray–white rectangle represents the clinical spectrum that narrows by the increase in the age of onset, where the gray beginning expresses the concurrence of black-and-white-indexed involvements.
[Source 269 ]Figure 23. Congenital erythropoietic porphyria (CEP)
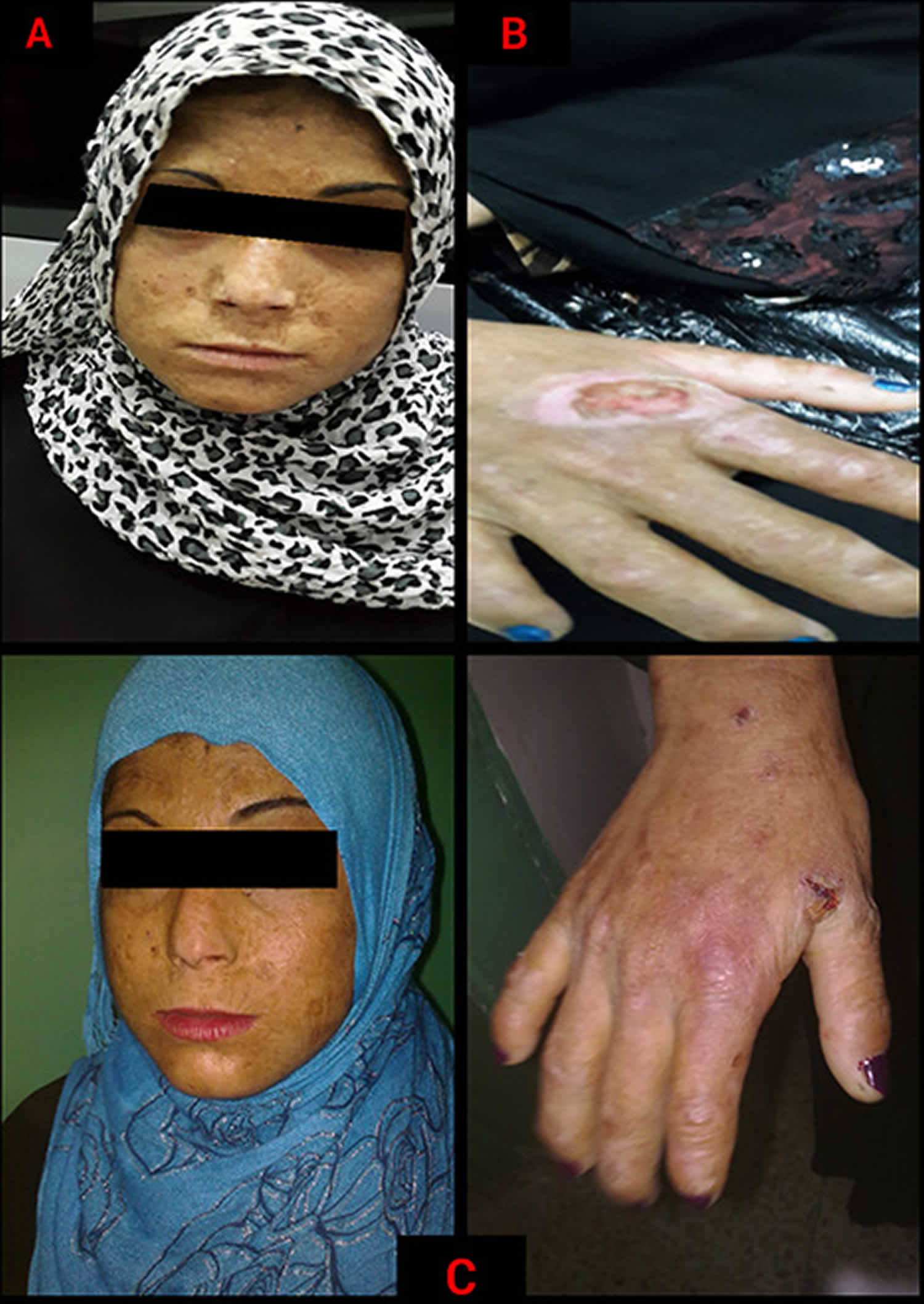
Footnotes: Congenital erythropoietic porphyria (CEP) in a 13-year-old female. (A) a dyspigmented sclerodermatous appearance of the face accompanied with linear and spotted atrophic lesions. (B) Immense areas of ulceration over the dorsum of the hand, with acrolysis of the bones and soft tissues at the ends of the fingers. (C) Healing ulcers on the face and dorsum of the hand after 1 month of treatment; the skin improved in terms of pigmentation and elasticity, with no active ulcers.
[Source 269 ]Figure 24. Congenital erythropoietic porphyria pathophysiology
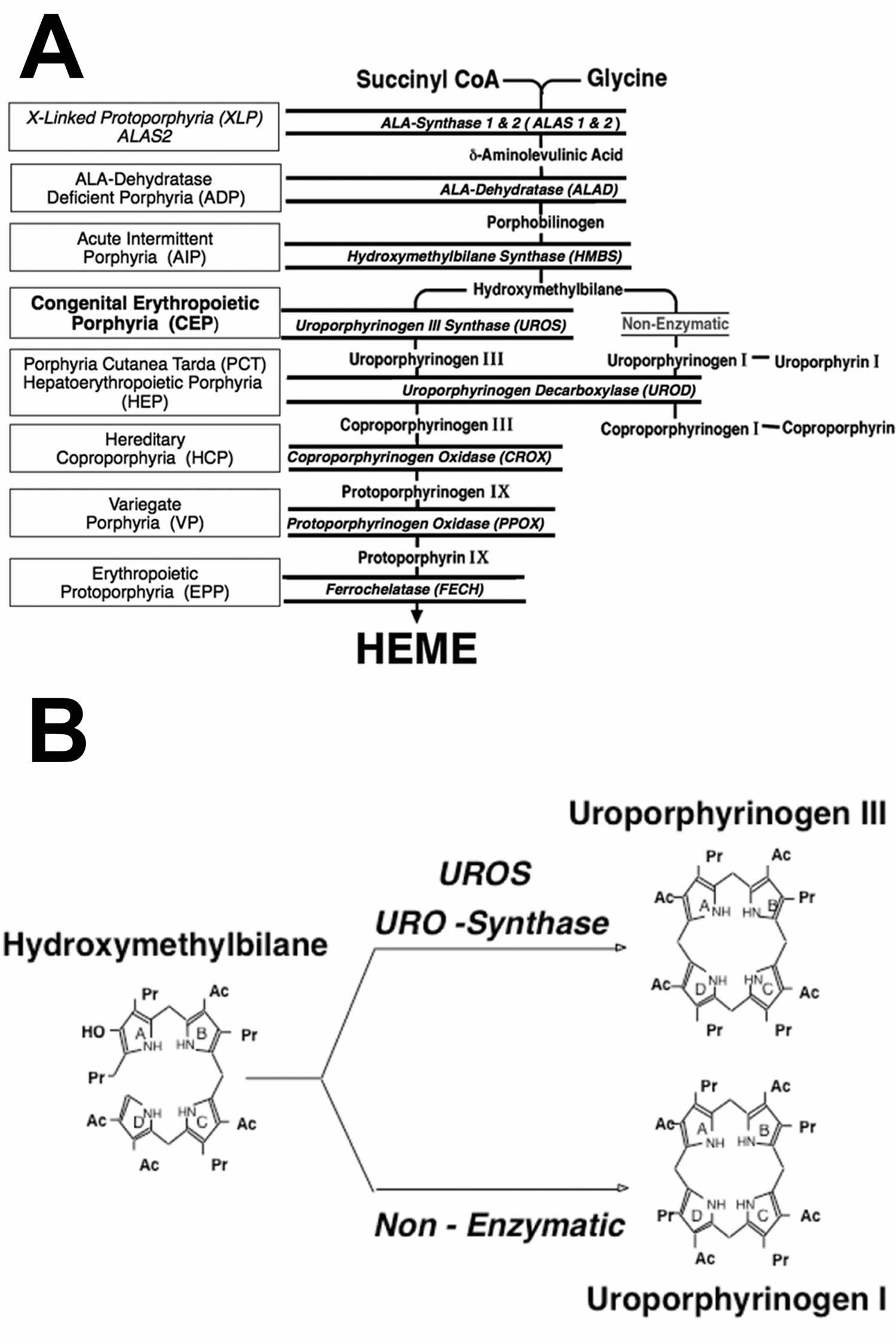
Foootnotes: (A) Heme biosynthetic pathway and the porphyrias resulting from the indicated heme biosynthetic enzyme defect. There are 8 enzymatic steps in the conversion of glycine and succinyl-CoA to heme. The heme biosynthetic enzymes are italicized, their substrates and products are indicated, and the resulting porphyrias are shown in boxes. Note that there are two aminolevulinic acid synthase (ALAS) isozymes: a housekeeping enzyme, ALAS1, encoded by a gene that is regulated by negative feedback repression by heme, and an erythroid specific enzyme, ALAS2, that is regulated by the iron response proteins and erythroid transcription binding proteins.
(B) Uroporphyrinogen synthase (UROS) normally converts hydroxymethylbilane (HMB) to uroporphyrinogen III. When the UROS activity is markedly decreased, HMB (hydroxymethylbilane) is non-enzymatically converted to uroporphyrinogen I, which is then enzymatically converted to oproporphyrinogen I by uroporphyrinogen decarboxylase (UROD). The accumulated uroporphyrinogen I and coproporphyrinogen I are oxidized to their respective porphyrins, which are photoactive and cause the sun/light-induced hemolysis and cutaneous manifestations of congenital erythropoietic porphyria (CEP).
[Source 275 ]Congenital erythropoietic porphyria causes
Congenital erythropoietic porphyria is caused by mutations in the UROS gene located on chromosome 10q26.2 284. Congenital erythropoietic porphyria (CEP) is rarely inherited in X-linked inheritance pattern due to mutation of the GATA1 gene located on the X chromosome (chromosome Xp11.23) that affected males only while the female heterozygotes were mostly asymptomatic 285, 274, 286, 281. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 274, 286. In a small percentage of congenital erythropoietic porphyria patients (>10%), a disease-causing mutation has not been detected in the UROS or GATA1 genes, raising the possibility that additional gene(s) may play a role in congenital erythropoietic porphyria pathogenesis 287.
Disease-causing mutations in either UROS gene or GATA1 gene result in absent or markedly reduced UROS (uroporphyrinogen III synthase) enzymatic activity. UROS (uroporphyrinogen III synthase) enzyme catalyzes the cyclization of hydroxymethylbilane (HMB) with concomitant inversion of ring D to yield uroporphyrinogen III (URO III) 288. Deficiency of UROS (uroporphyrinogen III synthase) enzyme results in accumulation of HMB (hydroxymethylbilane), most of which is converted nonenzymatically to non-physiological compound uroporphyrinogen I, which is then acted upon by uroporphyrinogen decarboxylase (UROD) the fifth enzyme in the heme synthetic pathway to form coproporphyrinogen I 280. Coproporphyrinogen I then oxidizes spontaneously to form the porphyrins coproporphyrin I and uroporphyrin I. In congenital erythropoietic porphyria (CEP), these toxic porphyrins mostly accumulate in red blood cell precursors in your bone marrow, as well as your teeth, bones, urine, and feces. These toxic porphyrins are not significantly elevated in your liver. Since these toxic porphyrins are photocatalytic compounds, exposure of the skin to sunlight and other sources of long-wave ultraviolet light elicits a phototoxic reaction, resulting in blistering and vesicle formation as well as increased friability of the skin and breakdown of red blood cells (hemolysis) 281, 289, 290.
Congenital erythropoietic porphyria inheritance pattern
Congenital erythropoietic porphyria (CEP) is inherited as an autosomal recessive genetic condition. Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. Patients with congenital erythropoietic porphyria (CEP) are either homozygotes or compound heterozygotes for mutations of the UROS gene at 10q25.2-q26.3 that encodes uroporphyrinogen III synthase (UROS), a cytosolic enzyme that converts hydroxymethyl bilane (HMB) to uroporphyrinogen III. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, and usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
Congenital erythropoietic porphyria (CEP) is rarely inherited in X-linked inheritance pattern due to mutation of the GATA1 gene located on the X chromosome that affected males, while the female heterozygotes were mostly asymptomatic 274, 286, 281. Females who are heterozygous for GATA1 gene will be either asymptomatic or have a milder disease with predominantly hematologic abnormalities due to skewed X-chromosome inactivation 274, 286.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 25. Congenital erythropoietic porphyria (CEP) autosomal recessive inheritance pattern
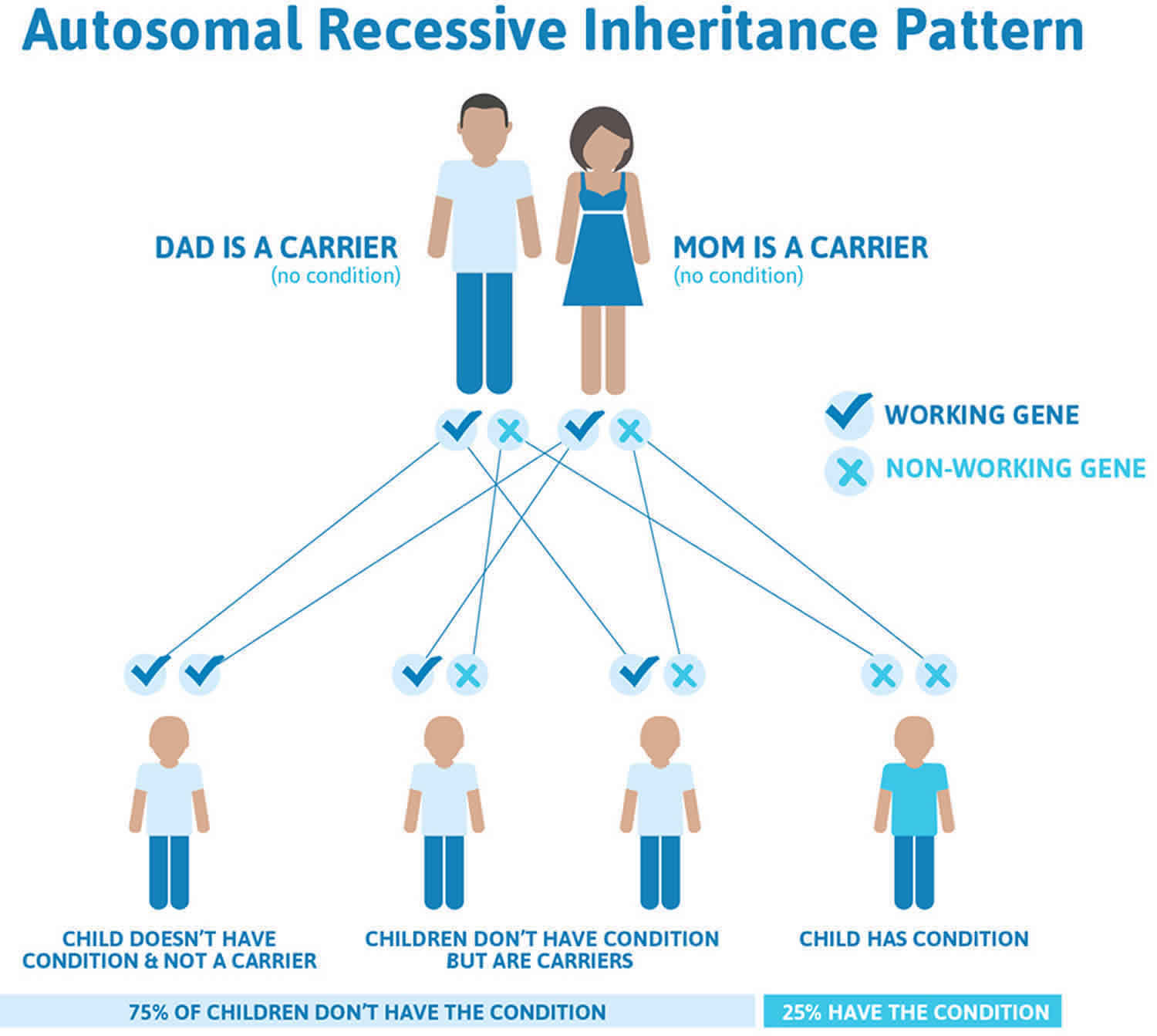
Congenital erythropoietic porphyria symptoms
Congenital erythropoietic porphyria or Gunther disease severity and age at onset of symptoms are highly variable between individuals. For many, the onset of symptoms and diagnosis are often in infancy. Some do not develop symptoms until adulthood, their symptoms are usually milder and primarily skin related 296.
Congenital erythropoietic porphyria symptoms usually start in infancy or childhood and the diagnosis in most patients is suggested by the reddish color of the urine which stains the diapers and fluoresces with a Wood’s lamp (ultraviolet light). Congenital erythropoietic porphyria (CEP) or Gunther disease major symptoms are sensitivity of the skin to sunlight and some types of artificial light, and anemia which can be very severe. Skin photosensitivity may be extreme, and can lead to blistering, severe scarring and increased hair growth (hypertrichosis). The fluid-filled blisters may rupture and get infected. These infected wound can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. Increased hair growth (hypertrichosis) on sun-exposed skin, brownish-colored teeth (erythrodontia), and reddish-colored urine are common. There may be bone fragility and vitamin deficiencies, especially vitamin D deficiency. Vitamin D is a fat-soluble vitamin that essential for bone development and maintenance, as it enhances calcium, magnesium, and phosphate absorption. Vitamin D deficiency due to sunlight avoidance in people with congenital erythropoietic porphyria can lead to a range of health problems, including bone disorders, muscle weakness, and an increased risk of fractures.
Red blood cells have a shortened life-span, and anemia often results. Synthesis of heme and hemoglobin are actually increased to compensate for the shortened red blood cell survival. The spleen can be enlarged due to extramedullary erythropoiesis, particularly in people with severe hemolytic anemia.
The most common symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of the skin to sunlight and some types of artificial light (photosensitivity), with blistering of the skin occurring after exposure. Affected individuals may also exhibit abnormal accumulations of body fluid under affected areas (edema) and/or persistent redness or inflammation of the skin (erythema). Affected areas of the skin may develop sac-like lesions (vesicles or bullae), scar, and/or become discolored (hyperpigmentation) if exposure to sunlight is prolonged. These affected areas of skin may become abnormally thick. In addition, in some cases, affected individuals may also exhibit malformations of the fingers and nails. The severity and degree of photosensitivity differ depending on the severity of the patient’s gene lesions which correlate with the deficient enzyme activity. In the great majority of congenital erythropoietic porphyria (CEP) cases, photosensitivity is seen from birth; however, in some cases, it may not occur until childhood, adolescence or adulthood. Patients also have brownish discolored teeth (erythrodontia), which fluoresce under ultraviolet light.
In more severe congenital erythropoietic porphyria (CEP) cases, other symptoms can include a low level of red blood cells (anemia), enlargement of the spleen (secondary hypersplenism), and increased hair growth (hypertrichosis) 275. The anemia can be severe and such patients require periodic blood transfusions to quickly increase the amount of red blood cells and iron in the blood. In severely affected patients, anemia may be present in the fetus (non-immune hydrops fetalis). Eye problems also can occur including corneal scarring, eye inflammation, and infections.
Skin involvement
Skin photosensitivity usually begins shortly after birth and is characterized by increased friability and blistering of the epidermis on the hands and face and other sun-exposed areas. Bullae and vesicles containing serous fluid which fluoresces due to their porphyrin content. Blisters are prone to rupture and become infected. Recurrent vesicle formation and secondary infection can lead to cutaneous scarring and deformities, as well as to loss of digits and facial features such as the eyelids, nose and ears. The skin may be thickened, with areas of hypo- and hyper-pigmentation and hypertrichosis of the face and extremities 297. Adult-onset congenital erythropoietic porphyria (CEP) patients have milder clinical symptoms and often exhibit only the skin manifestations of congenital erythropoietic porphyria (CEP) 298, 299, 300, 301, 302, 303. Photosensitivity symptoms are provoked mainly by visible light (400–410 nm Soret wavelength) and to a lesser degree by wavelengths in the long-wave UV region. Affected individuals are also sensitive to sunlight that passes through window glass that does not filter long-wave UVA or visible light as well as to light from artificial light sources 304.
Hemolytic anemia
Mild to severe hemolysis is accompanied by anisocytosis, poikilocytosis, polychromasia, basophilic stippling, reticulocytosis, increased nucleated red cells, absence of haptoglobin, increased unconjugated bilirubin and increased fecal urobilinogen. Plasma iron turnover also is increased 305. Hemolysis presumably results from the accumulated uroporphyrin I in erythrocytes. Secondary splenomegaly develops in response to the increased uptake of abnormal erythrocytes from the circulation, which may contribute to the anemia and also may result in leucopenia and thrombocytopenia. The latter is sometimes associated with significant bleeding and splenectomy may be beneficial in such cases. Anemia due to hemolysis can be severe. Erythroid hyperplasia and markedly ineffective erythropoiesis usually accompany hemolytic anemia in transfusion-dependent patients 306, 301, 307.
In the three male patients with congenital erythropoietic porphyria (CEP) due to GATA1 mutations, hematologic abnormalities including dyserythropoietic anemia, beta-thalassemia intermedia, thrombocytopenia, and hereditary persistence of fetal hemoglobin have been described 274, 286.
Other Clinical Features
Deposition of porphyrins can cause corneal ulcers and scarring, which can ultimately can lead to blindness 275. Other eye signs and symptoms can include scleral necrosis, necrotizing scleritis, seborrheic blepharitis, keratoconjunctivitis, sclerokeratitis, and ectropion 308, 309, 310. Porphyrins deposited in the teeth produce a reddish-brown color called erythrodontia. The teeth may fluoresce on exposure to long-wave ultraviolet light.
Deposition of porphyrins in bone causes osteopenia (lower than normal bone density) due to demineralization 311, 306, 312, 313. The risk for osteopenia and osteoporosis is further increased by vitamin D deficiency, which individuals with congenital erythropoietic porphyria (CEP) are prone to due to avoidance of sun exposure. Porphyrin accumulation in the bone can also cause expansion of the bone marrow, which can lead to hyperplastic bone marrow observed on biopsy 290, 297.
Congenital erythropoietic porphyria diagnosis
The diagnosis of congenital erythropoietic porphyria may be suspected when the reddish-colored urine is noted at birth or later in life. This finding, or the occurrence of skin blisters on sun or light exposure, should lead to a thorough clinical evaluation and specialized laboratory tests. The biochemical diagnosis of congenital erythropoietic porphyria is established by detection of markedly elevated levels of uroporphyrin I and coproporphyrin I in urine, red blood cells or amniotic fluid as well as high fecal coproporphyrin I concentrations. Diagnostic confirmation requires the demonstration of the specific UROS enzyme activity and/or by identifying the specific mutation(s) in the UROS gene which is/are responsible for the impaired enzyme 281.
Congenital erythropoietic porphyria should be considered in the differential diagnosis of non-immune hydrops fetalis, in which case the amniotic fluid surrounding the fetus will be pink, dark-red or brown, and should examined for porphyrins 314. Newborns with pink to dark-red urine-stained diapers should immediately have diagnostic studies. Congenital erythropoietic porphyria (CEP) also should be considered in children or adults who have porphyrinuria or skin blistering following exposure to sunlight or other sources of long-wave ultraviolet light. In some cases, the disease is less severe and presents in adult life with mild anemia and/or skin lesions.
Congenital erythropoietic porphyria (CEP) should be suspected in individuals with the following clinical and laboratory findings and family history 260.
- Nonimmune hydrops fetalis
- Signs of congenital erythropoietic porphyria
- Pink-to-dark red discoloration of the urine (pink or dark red urine-stained diapers are often the first sign in infants)
- Hemolytic anemia
- Severe cutaneous photosensitivity with onset usually in infancy or early childhood
- Blisters and vesicles in light-exposed areas, which are prone to rupture and infection
- Scarring and deformities (photomutilation) of digits and facial features, caused by recurrent blistering, infections, and bone resorption
- In light-exposed areas: friable skin, skin thickening, hypo- and hyperpigmentation
- Reddish-brown discoloration of teeth (fluoresce on exposure to long-wave ultraviolet light), also called erythrodontia
- Corneal ulcers and scarring
- Hypertrichosis of the face and extremities
- Laboratory findings include markedly increased levels of uroporphyrin I and coproporphyrin I isomers in red blood cells, urine, or amniotic fluid as well as coproporphyrin I in feces
Prenatal and preimplantation genetic diagnoses are available for subsequent pregnancies in congenital erythropoietic porphyria families.
Congenital erythropoietic porphyria treatment
There is no FDA-approved treatment for congenital erythropoietic porphyria (CEP) or specific treatment for the photosensitivity 260. Currently, the only effective management is prevention of skin blistering by strict avoidance of sun and light exposure, including the long-wave ultraviolet light that passes through window glass or is emitted from artificial light sources. Therefore, the use of protective clothing, wraparound sunglasses, protective window films, reddish incandescent bulbs, filtering screens for fluorescent lights, and opaque sunscreens containing zinc oxide or titanium oxide is recommended 260.
Wound care is necessary to prevent infection of opened blisters; surgical intervention may be necessary; blood transfusions with iron chelation are necessary when hemolysis is significant to maintain the hematocrit >35 to decrease reticulocytosis in transfusion-dependent patients 311, 260. Chronic transfusions have been useful in decreasing the bone marrow production of the phototoxic porphyrins.
Blood transfusions and perhaps removing the spleen may reduce porphyrin production by the bone marrow. Activated charcoal given by mouth is sometimes effective.
When successful, bone marrow transplantation (BMT) or hematopoietic stem cell (HSCT) transplantation has cured patients with congenital erythropoietic porphyria, but is accompanied by specific risks of complications and should be considered in children with severe skin and bone marrow involvement 260, 315, 316, 317, 318, 319, 320, 321, 322, 323, 324.
Evaluations Following Initial Diagnosis
To establish the extent of congenital erythropoietic porphyria disease and needs in an individual diagnosed with congenital erythropoietic porphyria (CEP), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Hematologic indices including reticulocytes and bilirubin (to assess hemolysis) and iron profile (to assess iron storage)
- Serum calcium and vitamin D concentrations; bone densitometry
- Liver function tests, especially in transfusion-dependent individuals given the risk for liver disease due to iron storage
- Skin evaluation to assess photosensitivity, photomutilation, and secondary skin changes (thickening, hyper- or hypopigmentation, hypertrichosis)
- Eye evaluation for corneal ulcers and scarring and other ocular manifestations
- Teeth assessment for erythrodontia (reddish-brown color from porphyrin deposition)
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of congenital erythropoietic porphyria (CEP) in order to facilitate medical and personal decision making
Monitoring
Monitor blood indices including iron profile, reticulocyte count, and bilirubin to assess hemolysis every six months. In those receiving blood transfusions, monitor for hemolysis more frequently and for iron overload. Monitor liver function and vitamin D 25-OH every six to twelve months in all affected individuals.
Agents to avoid
All affected individuals should avoid sunlight and UV light. To avoid eye complications and damage to the eyelids, wrap-around sun glasses should be worn. Corneal ulcers, scleritis, and blepharitis should be treated with topical antibiotics. In patients with ectropion, corrective surgery of the eyelid to help protect the cornea from injury may be indicated 322. In individuals undergoing surgeries, use of protective filters for artificial lights in the operating room to prevent phototoxic damage 255.
To avoid bone demineralization, vitamin D supplementation is indicated and bisphosphonates can be considered in individuals with osteoporosis 322.
In those with liver dysfunction avoid drugs that may induce cholestasis (e.g., estrogens).
Pregnancy Management
Successful pregnancies in women with congenital erythropoietic porphyria resulting in healthy and unaffected children have been described 325, 295.
Protective filters for artificial lights should be used in the delivery/operating room to prevent phototoxic damage to the mother during delivery 255.
Congenital erythropoietic porphyria prognosis
Congenital erythropoietic porphyria or Gunther disease severity and age at onset of symptoms are highly variable between individuals. For many, the onset of symptoms and diagnosis are often in infancy. Some do not develop symptoms until adulthood, their symptoms are usually milder and primarily skin related 296. Due to the hematological complications and an increased risk of infection, overall life expectancy may be markedly diminished in more severely affected patients 281. In addition, long-term damage, such as loss of fingers or toes and facial cartilage or contractures of the hands, can have a significant impact on patients’ quality of life, psychiatric well-being, and functional status with regard to daily life and ability to work.
Successful bone marrow (BMT) or hematopoietic stem cell (HSCT) transplantation is the only curative approach, but is accompanied by specific risks of complications and should be considered in children with severe skin and bone marrow involvement 260. The age of children with congenital erythropoietic porphyria (CEP) receiving bone marrow transplantation (BMT) ranges from younger than one year to 13 years 295. Some of the first individuals with congenital erythropoietic porphyria (CEP) to successfully undergo bone marrow transplantation in childhood are now in their 20s 316, 323. Although there is limited information available regarding their long-term outcome post receiving bone marrow transplantation (BMT), experts have learned that individuals successfully transplanted have essentially no photosensitivity to artificial light sources or sunlight.
Several cases of late-onset congenital erythropoietic porphyria (CEP) have been reported in patients older than 50 years of age who presented with skin congenital erythropoietic porphyria (CEP) symptoms and increased porphyrin metabolite excretion, though at a lower level than in patients with the classic infantile-onset congenital erythropoietic porphyria (CEP) presentation 275. In several cases, the occurrence of congenital erythropoietic porphyria (CEP) symptoms was associated with the presence of myelodysplastic syndrome (a group of blood cancers where the bone marrow doesn’t produce enough healthy blood cells leading to a deficiency in red blood cells, white blood cells, and platelets, causing symptoms like fatigue, increased risk of infection, and easy bruising or bleeding) and neither germline nor somatic mutations in UROS or GATA1 were detected [39–41]. However, low-level mosaicism in the bone marrow may not be picked up by sequencing methods, but may be sufficient to cause an accumulation of porphyrin metabolites resulting in an attenuated phenotype 326, 312, 327.
Recently, one 60 year-old male patient was described with late-onset skin porphyria symptoms and a urinary porphyrin metabolite pattern consistent with congenital erythropoietic porphyria (CEP). He was heterozygous for the common UROS gene mutation encoding p.C73R, but a second mutation was not detected. He did not have myelodysplastic syndrome or another underlying hematologic abnormality. While the exact pathophysiology was unclear in the absence of a second disease-causing mutation, acquired mosaicism in the bone marrow affecting the UROS gene was hypothesized to be the underlying cause 328.
Acute intermittent porphyria
Acute intermittent porphyria also called “AIP” or Swedish Porphyria is one of the acute porphyrias, which are a group of rare inherited metabolic disorders that involve defects in heme or ‘haem’ metabolism (a component of hemoglobin [Hb]) and result in excessive secretion of porphyrins and porphyrin precursors 329, 330, 331, 332, 333, 334, 335, 336, 337 , 49, 338. Heme or haem is a ring-shaped iron (Fe) containing molecule (organic compound containing an iron atom between the structure of the porphyrin ring) that commonly serves as a ligand of various proteins, more notably as a component of hemoglobin (the protein that carries oxygen in the blood), which is necessary to bind oxygen in the bloodstream and transport of oxygen in your body. Heme molecule also helps in respiration, detoxification of drugs, and other different biological functions. In hemoglobin, iron exists as ferrous (Fe2+) iron ion that is located at the center of the porphyrin ring, held in place by the four nitrogen atoms of the pyrrole rings (see Figures 1 to 3 below). Normally, your body makes heme (haem) in a multi-step process. Porphyrins are made during several steps of this process. People with porphyria are lacking certain enzymes needed for this process. This causes abnormal amounts of porphyrins or related chemicals to build up in the body. Porphyria occurs when the body cannot convert naturally occurring compounds called ‘porphyrins’ into heme. Porphyrins are substances that are required for the production of red blood cells. A common feature in all porphyrias is the accumulation in the body of porphyrins or porphyrin precursors. Although these are normal body chemicals, they normally do not accumulate. Precisely which of these chemicals builds up depends on the type of porphyria you have. There are 8 enzymes in the pathway for making heme and at least seven major forms of porphyria 339. The symptoms associated with the various forms of porphyria differ. It is important to note that people who have one type of porphyria do not develop any of the other types.
Porphyrias are generally classified into 2 groups: the “hepatic” (liver) and “erythropoietic” (red blood cell) types 339. Porphyrins and porphyrin precursors and related substances originate in excess amounts predominantly from the liver in the hepatic type porphyrias and mostly from the bone marrow in the erythropoietic type porphyrias. Porphyrias with skin manifestations are often referred to as “cutaneous porphyrias”. The term “acute porphyria” is used to describe porphyrias that can be associated with sudden attacks of pain and other neurological symptoms. The “hepatic” (liver) and “erythropoietic” (red blood cell) porphyrias can have cutaneous and acute symptoms, sometimes together. Most forms of porphyria are genetic inborn errors of metabolism.Acute intermittent porphyria (AIP) is an acute, hepatic or liver form of porphyria 339.
Acute intermittent porphyria (AIP) is caused by a deficiency in the enzyme porphobilinogen deaminase (PBGD) also known as hydroxymethylbilane synthase (HMBS). The porphobilinogen deaminase or hydroxymethylbilane synthase enzyme deficiency leads to a buildup of harmful substances called porphyrin precursors in your body, particularly in your liver. This enzyme deficiency is caused by a mutation in the HMBS gene which is inherited as an autosomal dominant trait (only one HMBS gene copy is affected). However, the enzyme deficiency by itself is not sufficient to produce symptoms of the disease and most individuals with a HMBS gene mutation do not develop symptoms of acute intermittent porphyria (AIP) 339. Additional factors such hormonal changes associated with puberty, the use of certain prescribed or recreational drugs, excess alcohol consumption, infections, and fasting or dietary changes are required to trigger the appearance of symptoms 339. Acute intermittent porphyria (AIP) symptoms include attacks of severe abdominal pain, constipation, a rapid heartbeat and increased blood pressure (tachycardia and hypertension), behavioral changes, seizures, and damage of the nerves to muscles (peripheral neuropathy) which can lead to complications like muscle weakness and paralysis. Moreover, unlike some other acute porphyrias, acute intermittent porphyria (AIP) does not manifest as sun sensitivity and skin rashes.
In Europe the prevalence of symptomatic acute intermittent porphyria (AIP) is reported to be 5.9 per million people in the general population 339. It is likely to be similar elsewhere in the world apart from Sweden where it is higher due to a founder effect. The founder effect is a type of genetic defect that occurs when a small group of individuals establishes a new population, leading to reduced genetic diversity compared to the original population 340. This can happen when a few individuals migrate from a larger population or when a population experiences a significant reduction in size. Over time, the resulting new subpopulation will have genetic and physical traits resembling the initial small, separated group, and these may be very different from the original larger population. Recent population based genetic studies have shown that approximately 1 in 2000 of the population inherit a disease causing (pathogenic) mutation in the HMBS gene. This suggests that only 1% of those who inherit the HMBS gene mutation will ever experience porphyria symptoms. Acute intermittent porphyria (AIP) can occur in individuals of all ethnic backgrounds, although it may be less frequently reported in African-American individuals. Women are affected by symptomatic acute intermittent porphyria (AIP) more often than men. The disorder is most common in young or middle-aged women. A study conducted in Sweden demonstrated an increased risk of schizophrenia or bipolar disorder in patients with acute intermittent porphyria (AIP), as well as in their relatives 341.
Clinical findings of an acute porphyria attack 330:
- Presence of otherwise unexplained severe, acute abdominal pain (without physical signs) in the vast majority (90%) of acute attacks 342. The pain, which occasionally may be more severe in the back or thighs, is usually only relieved with opiate analgesia. Atypical presentations are rare.
- During attacks nausea, vomiting, constipation, tachycardia, and hypertension are common.
- Muscle weakness, seizures, mental changes, and hyponatremia are features that alone or in combination increase the probability of acute porphyria.
- The urine may be reddish brown or red; however, this should not be used as a diagnostic criterion as it is not a constant finding, especially if the sample is fresh. The color is enhanced by exposure to air and light and reflects increased urinary concentrations of porphyrins and porphobilins formed from the porphyrin precursor porphobilinogen (PBG).
A diagnosis of acute intermittent porphyria (AIP) can be difficult because most symptoms are nonspecific and occur episodically. A diagnosis is usually based upon identification of characteristic symptoms from a detailed patient history, a thorough clinical evaluation and certain specialized tests. Acute intermittent porphyria (AIP) should be suspected in individuals with unexplained abdominal pain, especially repeated episodes and when occurring along with psychological symptoms, neurological findings with muscle weakness or unexplained hyponatraemia. Dark or reddish urine in such individuals is also suggestive of acute intermittent porphyria (AIP). However, absence of this feature does not exclude acute intermittent porphyria (AIP). The diagnosis of acute intermittent porphyria (AIP) can be confirmed by finding an elevated level of porphobilinogen (PBG) (> 6 mg/L) on a spot urine test during an acute attack 343. If the urinary concentration of porphobilinogen (PBG) is increased, molecular genetic testing is performed to confirm the diagnosis and/or to facilitate screening of family members. When a multigene panel or genomic testing has identified an HMBS gene mutation, the diagnosis of an intermittent porphyria (AIP) attack is confirmed when the urinary concentration of PBG is increased.
Acute intermittent porphyria (AIP) treatment is focused on preventing attacks by educating patients to avoid potential triggers (e.g, fasting, unsafe drugs). Acute attacks usually require hospital care and can be effectively treated with intravenous hematin.
Treatment options in acute intermittent porphyria (AIP) may include the following 343:
- Avoidance of precipitating factors (e.g, fasting, low caloric intake, high-risk porphyrogenic drugs, alcohol, infections and reproductive hormones change)
- High doses of glucose for mild attacks. Intravenous glucose (300 g/d in adults) is used in early acute attacks or when hemin is unavailable, but hemin infusion is more effective 344, 345.
- Hematin for severe attacks, especially those with severe neurologic symptoms. For sporadic acute neurovisceral attacks (i.e., when an individual has experienced one to ≤3 acute porphyria attacks in any 12-month period in the last two years): IV human hemin is the most effective treatment and may be lifesaving if employed early when neuronal damage is reversible. Hemin infusion rapidly downregulates delta-aminolevulinic acid synthase 1 (ALAS1) expression, decreases aminolevulinic acid (ALA) and porphobilinogen (PBG) accumulation, and resolves symptoms in 48–72 hour, although its effectiveness in preventing recurrent attacks is unclear 346, 347. If the criteria for recurrent attacks are met, Givlaari® (givosiran) should be considered, as long-term complications of hemin such as iron overload, phlebitis, and loss of venous access can be avoided. Alternative medical therapies to reduce frequency and/or severity of acute attacks when givosiran is not available include suppression of ovulation and prophylactic hemin infusion.
- During attacks, which generally last for several days, symptomatic treatment for pain and other manifestations (eg, tachycardia, nausea and vomiting, seizures)
- Gonadotropin-releasing hormone analogues for women with attacks related to their menstrual cycles
- Prophylactic hematin infusions
- Givosiran, in patients with acute hepatic porphyria, to decrease the rate of acute attacks 348
- Liver transplantation, as a last resort for patients with intractable recurrent attacks that are life-threatening or severely affect quality of life. Liver transplantation, as reported from several centers, is curative. Indications include repeated life-threatening acute porphyria attacks and poor quality of life when givosiran is not available or has shown insufficient medical efficacy.
Figure 26. Acute intermittent porphyria attack triggers
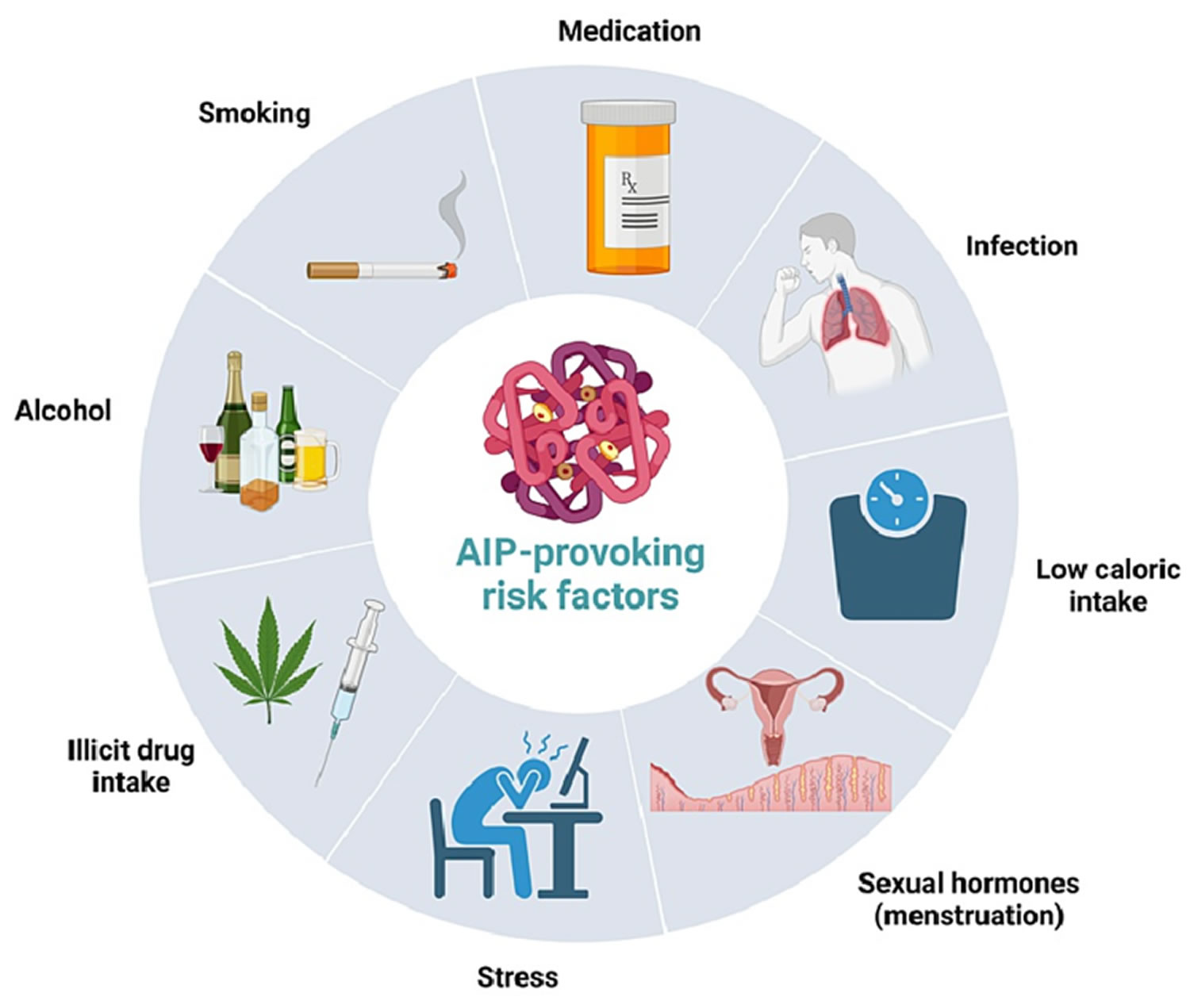
Figure 27. Acute intermittent porphyria signs and symptoms
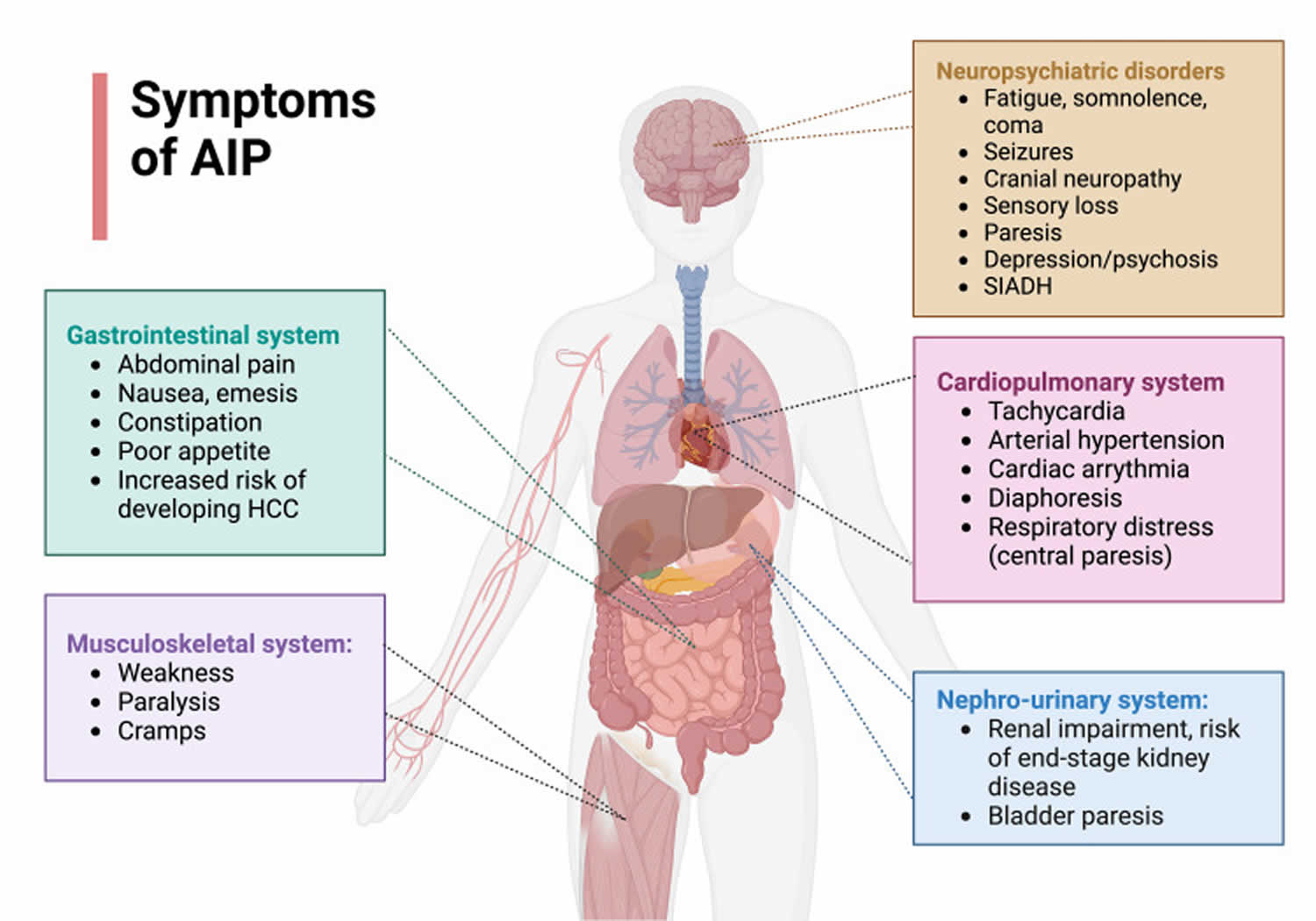
Abbreviations: AIP = acute intermittent porphyria; HCC = hepatocellular carcinoma; SIADH = syndrome of inappropriate antidiuretic hormone secretion
[Source 350 ]Acute intermittent porphyria cause
Acute intermittent porphyria (AIP) is a multifactorial disorder, which means that several different factors such as genetic and environmental factors occurring in combination are necessary for developing symptoms of the disorder 339. Individuals with acute intermittent porphyria (AIP) have a mutation in the HMBS gene located on the 11q24.1-q24.2 chromosome 351, 329. There are more than 500 mutations of the HMBS gene have been identified in the worldwide 336, 352. The HMBS gene provides instructions for making an enzyme known as hydroxymethylbilane synthase (HMBS) also known as porphobilinogen deaminase (PBGD) 353. The porphobilinogen deaminase (PBGD) or hydroxymethylbilane synthase (HMBS) enzyme is involved in the production of a molecule called heme. Heme is vital for all of your body’s organs, although it is most abundant in your blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). The production of heme is a multi-step process that requires 8 different enzymes. Hydroxymethylbilane synthase (porphobilinogen deaminase) is responsible for the third step in this process, which combines four molecules of porphobilinogen (the product of the second step) to form a compound called hydroxymethylbilane. In subsequent steps, five other enzymes produce and modify compounds that ultimately lead to heme.
Mutations in the HMBS gene lead to deficient levels of hydroxymethylbilane synthase (HMBS) or porphobilinogen deaminase (PBGD) in your body, which in turn can lead to the accumulation and release of porphyrin precursors, delta-aminolevulinic acid (ALA) and porphobilinogen (PBG) from the liver 354, 344.
However, the majority of people with a mutation in this gene do not develop symptoms of acute intermittent porphyria (AIP). Additional endogenous or exogenous factors, often called “triggers” are also required to cause symptomatic acute porphyria 339. These factors are not necessarily the same for each individual, and susceptibility to specific triggers may vary during a patient’s lifetime. Most of these triggers are believed to stimulate increased heme production (synthesis) in the liver and they include certain porphyrinogenic drugs, alcohol consumption, fasting or dieting (e.g. caloric restriction or low caloric intake), stress, infections or certain hormonal (endocrine) factors, female sex hormones, often in combination 339.
Acute intermittent porphyria is a low-penetrant genetic metabolic disease with penetrance considered to be around 10% to 20% 355. The penetrance of acute intermittent porphyria (AIP) in the general population has been estimated to be less than 1% 356. Manifest acute intermittent porphyria (MAIP) is considered when carriers develop typical acute neurovisceral attacks with an elevation of porphyrin precursors. In the absence of clinical episodes, it is referred to as latent acute intermittent porphyria (LAIP). Although higher penetrance has links to specific mutations, the overall genetic susceptibility factors underlying penetrance remain unknown.
Acute intermittent porphyria affects women to a greater degree than men, with a ratio of between 1.5 and 2 to 1. Attacks are rare before puberty. The typical age for the appearance of symptoms is between 18 to 40 years.
Symptomatic acute intermittent porphyria (AIP) is always accompanied by increased production and excretion of porphyrin precursors. However, for reasons that are unknown, some affected individuals have elevated porphyrin precursors without symptoms of acute intermittent porphyria (AIP). As discussed above, triggering factors are required for symptom development. The exact, underlying reasons why symptoms develop in some individuals with acute intermittent porphyria (AIP) are not fully understood. There are several theories as to the underlying pathogenesis of acute intermittent porphyria (AIP). One theory states that a specific porphyrin precursor (most likely 5-aminolevulinic acid [ALA]) is a neurotoxin that damages nerve tissue 339. This theory is supported by the information obtained from patients who have had liver transplant, which corrects both the clinical and biochemical features of the condition 339. A second theory suggests that heme deficiency in nerve cells (neurons) contributes to the development of symptoms 339. More research is necessary to determine the exact underlying mechanisms that are involved in the development of symptomatic episodes in individuals with acute intermittent porphyria (AIP).
Acute intermittent porphyria inheritance pattern
The HMBS gene mutation that predisposes individuals to developing acute intermittent porphyria (AIP) is inherited in an autosomal dominant pattern. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Dominant genetic disorders occur when only a single copy of an abnormal gene is sufficient for the appearance of the disease. The abnormal gene can be inherited from either parent, or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 28. Acute intermittent porphyria autosomal dominant inheritance pattern
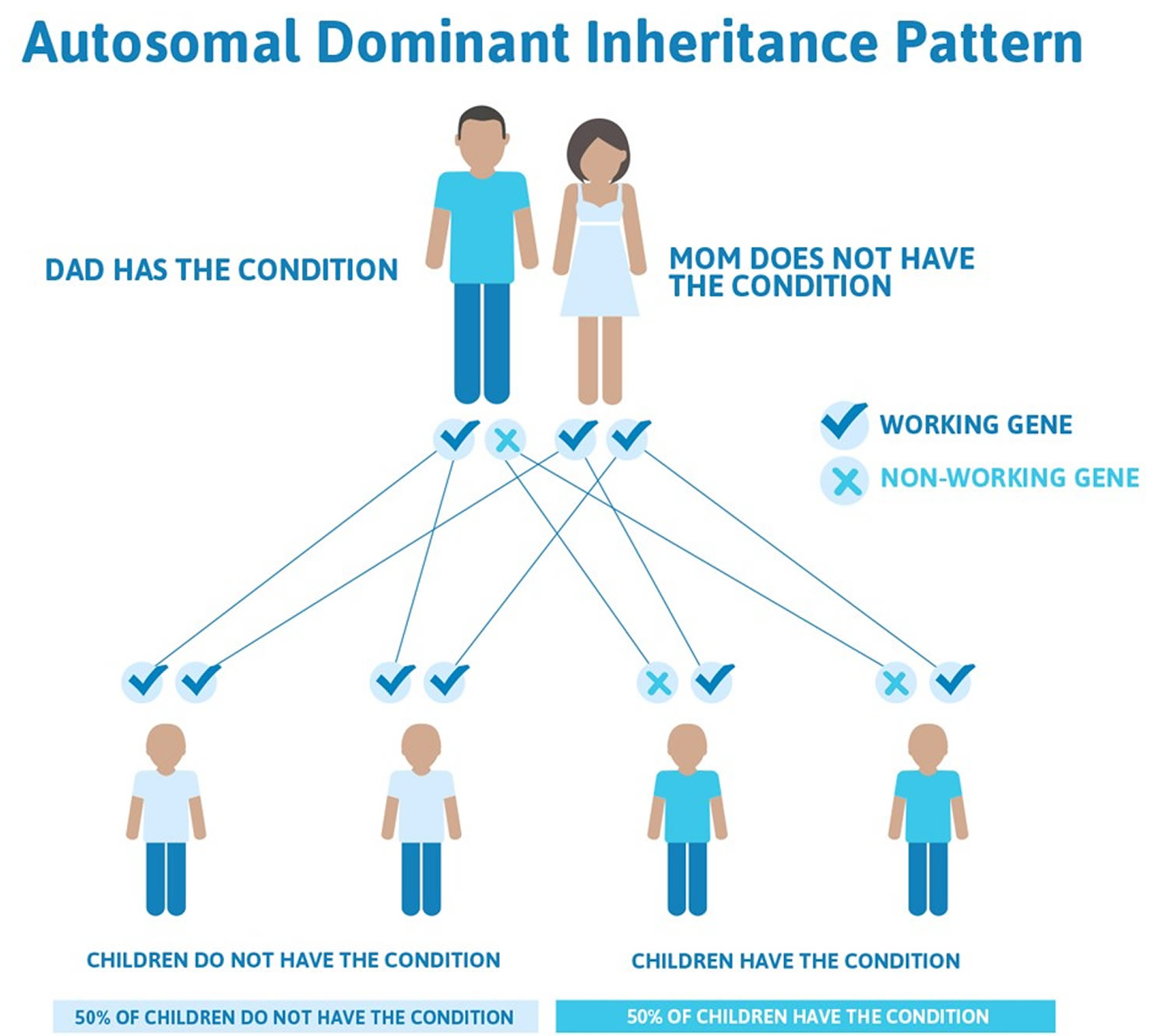
Acute intermittent porphyria pathophysiology
Acute intermittent porphyria (AIP) results from defects in the enzyme porphobilinogen deaminase (PBGD) also known as hydroxymethylbilane synthase (HMBS), which catalyzes the conversion of porphobilinogen (PBG) to hydroxymethylbilane. Impaired function of porphobilinogen deaminase (PBGD) leads to the accumulation of the porphyrin precursors porphobilinogen (PBG) and amino-levulinic acid (ALA). The predominant clinical problem appears to be neurologic damage that leads to peripheral and autonomic neuropathies and psychiatric manifestations 357.
Although levels of porphobilinogen (PBG) and amino-levulinic acid (ALA) are always elevated during acute attacks, how this leads to symptomatic acute intermittent porphyria is still unclear because most patients with the HMBS genetic defect have excessive porphyrin secretion but no symptoms.
In patients with acute intermittent porphyria (AIP), the function of porphobilinogen deaminase (PBGD) is only 40-60% of normal 329. With the advent of molecular technique, it has become clear that carriage of the HMBS genetic defect is much more common than symptomatic acute intermittent porphyria (AIP) 329. On average, out of 100 patients with the HMBS genetic defect, perhaps 10-20 secrete excess porphyrin precursors and only 1 to 2 have symptoms 329.
In acute intermittent porphyria (AIP), the neurologic damage occurs due to the accumulation of the porphyrin precursors, porphobilinogen (PBG) and aminolevulinic acid (ALA). The acute intermittent porphyria (AIP)-associated neurological damage manifests as peripheral and autonomic neuropathies and psychiatric manifestations.
A 2017 case-control study in 50 patients reported the association of acute intermittent porphyria (AIP) with systemic inflammation. Storjord et al. 358 found that the levels of insulin, C-peptide, prealbumin, and markers of kidney function, were decreased in symptomatic patients only, but not in asymptomatic ones. They postulated that in symptomatic patients of acute intermittent porphyria (AIP), the decrease in C-peptide levels in symptomatic acute intermittent porphyria (AIP) cases indicates that reduced insulin release is associated with enhanced disease activity and compromised kidney function 358.
Acute intermittent porphyria types
There 5 types of acute intermittent porphyria (AIP), caused by a heterozygous HMBS gene mutation and are based on the urine porphobilinogen (PBG)-to-creatinine ratio and occurrence of acute attacks 330, 359:
- Active (symptomatic) acute intermittent porphyria: An individual who has experienced at least one acute attack within the last two years.
- Sporadic AIP: 1-3 acute porphyria attacks in any 12-month period within the last 2 years
- Recurrent AIP: ≥4 acute porphyria attacks in a maximum period of 12 months within the last 2 years
- Symptomatic high excreter: Urine porphobilinogen (PBG)-to-creatinine ratio ≥4 times upper limit of normal (ULN) and no acute attacks in the last 2 years but has chronic long-standing manifestations of acute porphyria (e.g., pain or other porphyria-related manifestations in the absence of other likely explanations).
- Asymptomatic high excreter: Urine porphobilinogen (PBG)-to-creatinine ratio ≥4 times upper limit of normal (ULN) and no acute attacks in the last 2 years and no porphyria-related manifestations.
- Asymptomatic acute intermittent porphyria: Urine porphobilinogen (PBG)-to-creatinine ratio <4 times upper limit of normal (ULN) and no acute attacks in the last 2 years but has had ≥1 acute attack in the past.
- Latent (inactive) acute intermittent porphyria: Urine porphobilinogen (PBG)-to-creatinine ratio <4 times upper limit of normal (ULN) and no acute porphyria-related manifestations to date.
- Latent at-risk individual: An asymptomatic individual with a positive family history of acute intermittent porphyria (AIP) in whom an HMBS pathogenic variant was identified during screening of family members
- Latent low-risk individual: An asymptomatic individual with no known family history of acute intermittent porphyria (AIP) in whom the identification of an HMBS pathogenic variant was an incidental finding (the identification of a pathogenic variant in a gene that does not account for the phenotype that prompted the diagnostic testing).
Active (Symptomatic) acute intermittent porphyria
Active symptomatic acute intermittent porphyria, which are more common in women than men, are very rare before puberty 330. Onset typically occurs in the second or third decade 360.
The visceral, peripheral, autonomic, and/or central nervous systems (brain and spinal cord) may be affected, leading to a range of findings that are usually intermittent and sometimes life-threatening 330. The course of acute porphyria attacks is highly variable in an individual and between individuals 330.
Affected individuals may recover from acute porphyria attacks within days, but recovery from severe attacks that are not promptly recognized and treated may take weeks or months 330. Although attacks in most individuals are typically caused by exposure to certain endogenous or exogenous factors, it is not uncommon for individuals to have acute attacks in which no precipitating factor can be identified 330.
Acute porphyria attacks
An acute porphyria attack is defined as an episode that includes significantly increased urinary porphobilinogen (PBG) concentration and two or more of the clinical manifestations of an acute porphyria attack that typically persist for more than 24 hours in the absence of other likely explanations 361.
Severe abdominal pain, which may be generalized or localized and not accompanied by muscle guarding, is the most common symptom and is often the initial sign of an acute attack. Back, buttock, or limb pain may be a feature. Gastrointestinal features including nausea, vomiting, constipation or diarrhea, and abdominal distention are common, and ileus can occur. Tachycardia and hypertension are frequent, while fever, sweating, restlessness, and tremor are seen less frequently. Urinary retention, incontinence, and dysuria may be present.
Approximately 3%-8% of individuals with acute intermittent porphyria (AIP), mainly women, experience recurrent acute intermittent porphyria (AIP) (defined as ≥4 attacks in one year) for a prolonged period, often many years 361, 342.
Acute porphyria triggers
Acute porphyria attacks may be precipitated or triggered by endogenous or exogenous factors. These include the following 362:
- Prescribed and recreational drugs that are detoxified in the liver by cytochrome P450 and/or result in induction of 5-aminolevulinic acid (ALA) synthase and heme biosynthesis. Prescription drugs that can precipitate an attack include, for example, barbiturates, sulfa-containing antibiotics and antibiotics for urinary tract infections, some anti-seizure medications, progestogens, and synthetic estrogens (see a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/).
- Endocrine factors. Reproductive hormones play an important role in the clinical expression of acute intermittent porphyria (AIP). In women, acute neurovisceral attacks related to the menstrual cycle, usually the luteal phase, are common 362. Pregnancy in women with acute intermittent porphyria (AIP) is usually uncomplicated, and although urinary PBG concentration may increase during pregnancy, this does not lead to a higher frequency of clinical porphyria manifestations 363. However, there is a higher risk for pregnancy-induced hypertensive disorder, gestational diabetes, and infants with intrauterine growth restriction. In general, risk ratios are higher among women with acute intermittent porphyria (AIP) who have high lifetime urinary porphobilinogen (PBG) concentrations 364.
- Fasting. A recognized precipitating factor is inadequate caloric intake in connection with, for example, dieting or heavy exercise schedules 362.
- Stress. Psychosocial and other stresses, including intercurrent illnesses, infections, alcoholic excess, and surgery, can precipitate an attack 365
Peripheral neuropathy
Peripheral neuropathy is predominantly motor and is less common now than in the past, due to the availability of better treatments that reduce the risk of long duration of untreated acute porphyria attacks, the main risk factor for neurologic manifestations and long-term neurologic complications 330. Muscle weakness often begins proximally in the legs but may involve the arms or legs distally and can progress to include respiratory muscles, resulting in complete paralysis with respiratory failure 330. Bilateral axonal motor neuropathy may also involve the distal radial nerves. Motor neuropathy may also affect the cranial nerves or lead to bulbar paralysis.
Patchy sensory neuropathy may also occur 330.
Central nervous system signs
Mild mental changes such as anxiety, insomnia, irritability, and even mild cognitive impairment occur in up to 80% of symptomatic individuals and often in the initial stages of an acute porphyria attack 342.
Severe mental symptoms attributed to acute encephalopathy characterize the severe acute porphyria attack, manifesting as aberrant behavior, hallucinations, confusion, impaired consciousness, or seizures 366, 361.
Brain MRI changes can be detected in 47% of individuals with severe mental changes, usually in the form of posterior reversible encephalopathy syndrome, but normal MRI examination despite acute encephalopathy also occurs 366.
Hyponatremia is present in 25%-61% of acute porphyria attacks due to sodium loss, overhydration, hypothalamic involvement (i.e., syndrome of inappropriate antidiuretic hormone [SIADH]), or a combination of these conditions 366, 367.
Seizures occur in 1%-20% of acute porphyria attacks, with or without hyponatremia. They are transient and only present in severe attacks with acute encephalopathy; they do not occur in remission 366.
High Urine Porphobilinogen (PBG) Excreter
- Symptomatic high excreter. An individual with permanently high urinary porphobilinogen (PBG) concentration is considered symptomatic (i.e., a symptomatic high excreter) when having porphyria-related manifestations, usually pain, peripheral neuropathy, and psychiatric symptoms. Management of these manifestations is based on the need for supportive drugs, all of which should be evaluated for safety in acute intermittent porphyria (AIP). The condition usually occurs after an acute porphyria attack and can persist for many years 368. In a Swedish acute intermittent porphyria (AIP) cohort approximately 10% of adults with acute intermittent porphyria (AIP) are high urinary porphobilinogen (PBG) excreters 369.
- Asymptomatic high excreter. An asymptomatic HMBS heterozygote with permanently high urinary porphobilinogen (PBG) concentration (urine PBG-to-creatinine ratio ≥4 times the upper limit of normal) and has had no porphyria-related manifestations during the last two years.
Asymptomatic acute intermittent porphyria
Asymptomatic acute intermittent porphyria also called acute porphyria in remission refers to a person has had one or more acute porphyria attacks in the past but has had no acute porphyria-related manifestations during the last two years and has a urine porphobilinogen (PBG)-to-creatinine ratio is less than four times the upper limit of normal.
Latent acute intermittent porphyria
Latent acute intermittent porphyria refers to a person who is heterozygous for an HMBS pathogenic variant associated with acute intermittent porphyria who has never experienced acute porphyria-related manifestations and does not have significantly elevated urinary porphobilinogen (PBG) concentration.
The risk of becoming symptomatic depends on age, sex, and exposure to provoking agents, and is higher if the individual belongs to a family with other symptomatic individuals 370.
Acute intermittent porphyria signs and symptoms
Most people who inherit the HMBS gene for acute intermittent porphyria (AIP) never develop symptoms. However, experts recommend that all relatives of someone with acute intermittent porphyria obtain testing, to determine who has the genetic trait and who does not. Those who test positive for the trait should be educated as to measures that will help avoid attacks. Prevention is essential to good management.
It is important to note the highly variable nature of acute intermittent porphyria (AIP) and that affected individuals may not have all of the symptoms discussed below. Acute intermittent porphyria (AIP) can be associated with a range of symptoms and physical findings that can potentially involve multiple organ systems of the body. The course and severity of attacks is highly variable from one person to another. In some cases, particularly those without proper diagnosis and treatment, acute intermittent porphyria (AIP) can potentially cause life-threatening complications. Affected individuals and parents of affected children should talk to their physician and medical team about their specific case, associated symptoms and overall prognosis.
Acute intermittent porphyria (AIP) rarely attacks before puberty, with typical symptoms occurring between the ages of 20 and 40, more in females than males due to female sex hormones 332. Symptoms usually come as discrete attacks that develop over two or more days.
The abdominal pain is typically severe, epigastric, and colicky and can be severe and occurs in most cases. It tends to last for several days. The abdominal pain can be associated with constipation, nausea and vomiting 330.
Other symptoms may include:
- nausea
- vomiting
- constipation
- pain in the back, arms and legs
- muscle weakness (due to effects on nerves supplying the muscles)
- urinary retention
- palpitation (due to a rapid heart rate and often accompanied by increased blood pressure)
- Sometimes red or brown urine due to elevated porphobilinogen (PBG) in urine, which are the immediate precursors proximal to the hydroxymethylbilane synthase (HMBS), may be observed, which darkens on exposure to air, light, and warmth 371.
Central nervous system signs may include delirium, confusion, hallucinations, weakness with progression to quadriplegia and respiratory failure, cortical blindness, and even coma. In 5% of cases, patients can develop seizures, with partial seizures being the most common subtype 372
Sometimes the level of salt (sodium and chloride) in the blood decreases markedly and contributes to some of these symptoms. The skin is not affected.
The symptoms of acute intermittent porphyria (AIP) usually occur as episodes or “attacks” that develop over course of several hours or a few days. Affected individuals usually recover from an attack within days. However, if an acute attack is not diagnosed and treated promptly recovery can take much longer, even weeks or months. Most affected individuals do not exhibit any symptoms in between episodes. Onset of attacks usually occurs in the 20s or 30s, but may rarely occur at or just after puberty. Onset before puberty is extremely rare. Attacks are much more common in women than men, probably because of the menstrual cycle hormones. Approximately 3%-5% of affected individuals, predominately women, experience recurrent attacks, which are defined as more than 4 per year, for a period of many years.
Abdominal pain, which is usually severe, is the most common symptom associated with acute intermittent porphyria (AIP) and often the initial sign of an attack. Abdominal pain is usually severe, steady (unremitting) and widespread (diffuse). Less often, abdominal pain is described as cramping. Pain may also occur in the neck, lower back, buttocks, or arms and legs.
Gastrointestinal symptoms are also common during an attack and can include nausea, vomiting, constipation or diarrhea, and abdominal swelling (distention). A painful blockage or obstruction (ileus) of part of the small intestines may also occur. Difficulty passing urine (urinary retention) can also occur.
Neurological symptoms may also develop including damage to the nerves outside the central nervous system (peripheral neuropathy). Peripheral neuropathy is characterized by numbness or tingling and burning sensations that usually begin in the feet and sometimes the arms. Affected individuals may develop muscle weakness in the legs that may progress to affect the arms and the trunk of the body, eventually causing partial loss or impairment of motor function (motor paralysis). In rare cases, the muscles used to breathe can become involved and potentially cause life-threatening respiratory failure which requires mechanical ventilation.
During attacks some individuals develop psychological symptoms including irritability, depression, anxiety, insomnia, hallucinations, paranoia, disorientation, and altered consciousness ranging from excessive drowsiness (somnolence) to agitation or, in severe cases, coma.
Affected individuals may also experience a faster than normal heart rate (tachycardia) , high blood pressure (hypertension) and irregular heartbeats (cardiac arrhythmias). Seizures have also been reported. Abnormally low sodium levels (hyponatremia) may develop rapidly during an attack and contribute to the onset of seizures.
Patients have been reported to be completely free of symptoms in between the attacks. However, it is also suggested that 20% to 64% of patients may suffer from disabling chronic signs and symptoms such as pain, nausea, fatigue, and neuropathic features, including numbness and tingling sensations 373.
Individuals with chronic acute intermittent porphyria (AIP) may also develop complications that occur after many years (long-term complications) such as high blood pressure (hypertension), kidney damage potentially resulting in kidney failure, and liver cancers such as hepatocellular carcinoma (liver cancer) or cholangiocarcinoma (bile duct cancer).
A study conducted in Sweden demonstrated an increased risk of schizophrenia or bipolar disorder in patients with acute intermittent porphyria (AIP), as well as in their relatives 341.
Acute intermittent porphyria complications
Acute intermittent porphyria complications include:
- Systemic arterial hypertension and chronic kidney disease – Reports exist of the prevalence of up to 30% of these closely linked disorders in patients with acute intermittent porphyria (AIP) 374. End-stage renal disease (ESRD) is a life-threatening complication in acute intermittent porphyria (AIP) patients with chronic active disease 375
- Muscle denervation is another major pathologic complication 376. Some patients after acute attacks have residual deficits such as foot/wrist drop or wasting of the intrinsic muscles of hands 377.
- Hepatocellular carcinoma (liver cancer): This is the most deadly long-term complication of acute intermittent porphyria (AIP). Results of studies performed over the last 3 to 4 decades have shown a remarkably increased incidence of hepatocellular carcinoma (liver cancer) in acute intermittent porphyria (AIP) patients compared with the general population 378. A Swedish study showed that the risk for hepatocellular carcinoma (liver cancer) in AIP is increased 80 times after the age of 50 years 379, 330. It is worthwhile to know that acute intermittent porphyria (AIP) -associated hepatocellular carcinoma (liver cancer) is typically free from the usual preceding comorbidities such as Hepatitis B or C infection.
Acute intermittent porphyria diagnosis
A diagnosis of acute intermittent porphyria (AIP) can be difficult because most symptoms are nonspecific and occur episodically. A diagnosis is usually based upon identification of characteristic symptoms from a detailed patient history, a thorough clinical evaluation and certain specialized tests. Acute intermittent porphyria (AIP) should be suspected in individuals with unexplained abdominal pain, especially repeated episodes and when occurring along with psychological symptoms, neurological findings with muscle weakness or unexplained hyponatraemia. Dark or reddish urine in such individuals is also suggestive of acute intermittent porphyria (AIP). However, absence of this feature does not exclude acute intermittent porphyria (AIP). The diagnosis of acute intermittent porphyria (AIP) can be confirmed by finding an elevated level of porphobilinogen (PBG) (> 6 mg/L) on a spot urine test during an acute attack 343. If the urinary concentration of porphobilinogen (PBG) is increased, molecular genetic testing is performed to confirm the diagnosis and/or to facilitate screening of family members. When a multigene panel or genomic testing has identified an HMBS gene mutation, the diagnosis of an intermittent porphyria (AIP) attack is confirmed when the urinary concentration of PBG is increased.
Clinical Testing and Workup
Screening tests to measure the levels of the porphyrin precursor porphobilinogen (PBG) in urine are essential to confirm a diagnosis of acute porphyria. Acute attacks are always accompanied by increased production and excretion of porphyrin precursor porphobilinogen (PBG) in acute intermittent porphyria (AIP). If urinary porphobilinogen (PBG) excretion is increased, then further testing (fecal and blood porphyrin measurement) is necessary to distinguish acute intermittent porphyria (AIP) from variegate porphyria or hereditary coproporphyria. This should not delay treatment of acutely unwell patients. Delta-aminolevulinic acid (ALA) excretion will also be elevated in urine samples from individuals with acute intermittent porphyria (AIP), but measurement is less widely available and is not essential. These tests can be performed on a random (spot) urine sample that should be protected from light after collection and during transport to the laboratory. There is now good evidence that once urine porphobilinogen (PBG) excretion is increased in acute intermittent porphyria (AIP) it takes many years to return to normal. Increased urine porphobilinogen (PBG) excretion in a known acute intermittent porphyria (AIP) patient does not therefore prove that a patient is having an acute attack.
Family Testing
Molecular genetic testing is not essential to confirm a diagnosis as the porphyrin biochemical findings are characteristic. However molecular genetic testing to detect a mutation in the HMBS gene is usually required so that family members can be offered testing for this mutation. Genetic testing is available mainly from laboratories specializing in porphyria diagnosis.
Patients and family members who have inherited acute intermittent porphyria (AIP) should be advised on how to limit their risk of any future acute attacks. This should include information about acute intermittent porphyria (AIP) and what causes attacks, how to check if a prescribed medication is safe or unsafe and details of relevant patient support groups.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Evaluations Following Initial Diagnosis of Acute Intermittent Porphyria (AIP) Attack
To establish the extent of disease and needs in an individual diagnosed with acute intermittent porphyria (AIP) who is experiencing acute signs and symptoms, the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Consider other causes of abdominal pain in addition to porphyria.
- Review all medications and discontinue any that can exacerbate acute porphyria 380.
- Initial investigations should include the following:
- Complete blood count
- Measurement of serum/plasma concentrations of urea, creatinine, and electrolytes
- If there is hyponatremia, measure serum and urine osmolality, and urine sodium concentration
- Other blood tests as indicated by the individual’s condition and possible cause of the attack (e.g., C-reactive protein, blood cultures, serum creatine kinase, and plasma magnesium concentration)
- Brain MRI when central nervous system manifestations are present
- Refer individual to a porphyria specialist for more detailed clinical advice on acute intermittent porphyria (AIP).
- Affected individuals should be advised to register with an organization that provides warning jewelry in case of an accident (e.g., MedicAlert® or similar).
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse is recommended to inform affected individuals and their families about the nature, mode of inheritance, and implications of AIP to facilitate medical and personal decision making.
- Assess need for family support and resources including community or online resources and home nursing referral.
Acute intermittent porphyria differential diagnosis
Symptoms of the following disorders can be similar to those of acute intermittent porphyria (AIP). Comparisons may be useful for a differential diagnosis.
The acute attacks that characterize acute intermittent porphyria (AIP) are similar to those seen in 3 other forms of porphyria specifically variegate porphyria, hereditary coproporphyria, and ALA-Dehydratase deficiency porphyria. Collectively, these four forms of the porphyria are classified as the acute porphyrias.
- Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP) is the most common form of Guillain-Barré Syndrome (GBS), an that affects the peripheral nervous system. Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP) occurs when the body’s immune system mistakenly attacks the myelin sheath, the protective covering of the nerves, leading to nerve damage and impaired nerve signaling. It is characterized by rapidly progressive weakness and sensory loss in the limbs due to inflammation and damage to the myelin sheath of peripheral nerves.
- Miller Fisher syndrome (MFS) is a rare neurological disorder considered a variant of Guillain-Barré syndrome (GBS) 383. Miller Fisher syndrome (MFS) is characterized by a triad of symptoms: ataxia (loss of coordination), areflexia (loss of reflexes), and ophthalmoplegia (paralysis of the eye muscles). While MFS is rare, affecting 1-2 people per million each year, it is typically self-limiting, with most individuals recovering within 6 months even without specific treatment
- Acute Motor-Sensory Axonal Neuropathy (AMSAN) is a severe variant of Guillain-Barré syndrome (GBS) characterized by rapidly progressive weakness and sensory loss, often requiring ventilation and having a prolonged recovery period 384. Acute Motor-Sensory Axonal Neuropathy (AMSAN) is an axonal subtype, meaning it primarily affects the nerve fibers (axons) rather than the myelin sheaths that surround them.
- Acute motor axonal neuropathy (AMAN) is a rare variant of Guillain-Barré syndrome (GBS) characterized by acute, predominantly motor paralysis, with minimal or no sensory loss 385. Acute motor axonal neuropathy (AMAN) is an autoimmune disorder where the body’s immune system attacks the motor axons (nerve fibers). Acute motor axonal neuropathy (AMAN) is often associated with prior infection, particularly by Campylobacter jejuni, and can lead to significant motor weakness and, in some cases, respiratory failure.
Causes of acute abdomen – Peritonitis, appendicitis, acute cholecystitis, acute gastritis, acute pancreatitis, intestinal occlusion, strangulated abdominal hernia, acute mesenteric ischemia, ileus, diverticulitis, esophagitis, endometriosis, gastric outlet obstruction, intussusception, pelvic inflammatory disease, ovarian cysts, acute pyelonephritis, aortic dissection.
Tyrosinemia type 1 is a rare autosomal recessive genetic metabolic disorder characterized by lack of the enzyme fumarylacetoacetate hydrolase (FAH), which is needed for the final break down of the amino acid tyrosine. Failure to properly break down tyrosine leads to abnormal accumulation of tyrosine and its metabolites in the liver, including a heme precursor aminolevulinic acid (ALA), potentially resulting in severe liver disease. Tyrosine may also accumulate in the kidneys and central nervous system. Symptoms and physical findings associated with tyrosinemia type 1 appear in the first months of life and include failure to gain weight and grow at the expected rate (failure to thrive), fever, diarrhea, vomiting, an abnormally enlarged liver (hepatomegaly), and yellowing of the skin and the whites of the eyes (jaundice). Tyrosinemia type 1 may progress to more serious complications such as severe liver disease, cirrhosis, and hepatocellular carcinoma if left untreated. Untreated children can also suffer neurological crises similar to those seen in acute porphyria. Treatment with nitisinone and a low-tyrosine diet should begin as soon as possible after the diagnosis is confirmed.
Lead toxicity can cause symptoms that mimic acute porphyria (acute abdominal pain, constipation, neuropathy). Lead inhibits several of the enzymes of heme biosynthesis, which can therefore result in an increase in urine coproporphyrin and 5-aminolevulinic acid excretion, but not porphobilinogen excretion. It can also cause an increase in erythrocyte protoporphyrin concentration, although this is all the zinc-chelate form (zinc-protoporphyrin). The definitive test for lead poisoning is blood lead measurement.
Acute intermittent porphyria treatment
The treatment of acute intermittent porphyria (AIP) is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, neurologists, hematologists, hepatologists, psychiatrists, and other healthcare professionals may need to systematically and comprehensively plan an affected patient’s treatment. Genetic counseling may benefit affected individuals and their families.
The objective of acute intermittent porphyria (AIP) treatment is to manage your symptoms, prevent complications and to suppress heme synthesis in your liver with hematin, which reduces the production of porphyrin precursors. Initial treatment steps also include stopping any medications that can potentially worsen acute intermittent porphyria (AIP) or cause an attack and ensuring proper caloric intake, which can include intravenous infusion of sufficient nutrients (glucose and salt). Carbohydrate loading in conjunction with good pain medication may be sufficient for mild attacks.
Hospitalization is often necessary for acute attacks, particularly if nausea and vomiting have prevented adequate oral intake. Medications for pain, nausea and vomiting, intravenous (IV) hydration, and close observation are generally required.
In the United States, affected individuals may be treated with Panhematin (hemin for injection), an enzyme inhibitor derived from red blood cells that is potent in suppressing acute attacks of porphyria. Panhematin almost always returns porphyrin and porphyrin precursor levels to normal values. The U.S. Food and Drug Administration (FDA) approved Panhematin for the treatment of recurrent attacks of acute intermittent porphyria (AIP) related to the menstrual cycle in susceptible women, after a trial of glucose therapy and should be administered only by physicians experienced in the management of porphyrias in a hospital setting. Based on much experience, it is used for treating and even preventing acute attacks, often without an initial trial of glucose, and has been found to be safe during pregnancy.
Glucose and other carbohydrates can help suppress disease activity, are given by vein or by mouth, and are part of initial treatment. Intravenous heme, however, is both more specific and effective than glucose and should be started if the patient’s symptoms fail to improve within 36 hours. Heme is sold as Panhematin®, from Recordati Rare Diseases (https://recordatirarediseases.com/products). Most hospitals do not stock it. Therefore the pharmacy must be notified at the time the patient’s admission to initiate a request for air-freighting enough medication for 5 days of treatment. Generally, shipping will take at least 24 hours.
Panhematin, is the only commercially available form of heme for treatment and prevention of acute porphyric attacks in the United States. Heme arginate, which is marketed in other countries as Normosang® (Orphan Europe), is another preparation for intravenous administration. The main side-effect of Panhematin® is irritation of the vein used for infusion (phlebitis). This is avoided by slow infusion through a large caliber vein or central line. Adding human albumin to the heme solution also may reduce the risk of phlebitis. (Directions for preparing Panhematin® in this manner can be obtained from porphyria specialist and is included in the Primary Care Physician/Emergency Room Kit.) Heme therapy is indicated only if an acute attack of porphyria is proven by a marked increase in urine porphobilinogen. It may be useful also as preventive therapy for people with frequent recurrent attacks.
In 2019, the FDA approved Givlaari (givosiran) for the treatment of adult patients with acute hepatic porphyria, including acute intermittent porphyria (AIP). Givlaari (givosiran) aims to reduce the number of attacks patients experience.
Normosang (heme arginate) is another heme preparation that can be used to treat individuals with acute intermittent porphyria (AIP). Normosang is not available in the United States, but is used in many other countries where Panhematin is not available.
Treatment for acute intermittent porphyria (AIP) also includes drugs to treat specific symptoms such as certain pain medications (analgesics), anti-anxiety drugs, anti-hypertensive drugs, and drugs to treat nausea and vomiting, tachycardia, or restlessness. The pain is usually very severe and generally requires opiates (e.g. morphine) for adequate relief. Medications to treat any infections that may occur at the same time as an attack (intercurrent infection) may also be necessary. Although many types of drugs are believed to be safe in individuals with acute intermittent porphyria (AIP), recommendations about drugs for treating acute intermittent porphyria (AIP) are based upon experience and clinical study. Since many commonly used drugs have not been tested for their effects on porphyria, they should be avoided if at all possible. If a question of drug safety arises, a physician or medical center specializing in porphyria should be contacted. A list of these institutions may be obtained from the American Porphyria Foundation. The Foundation also maintains an Acute Porphyria Drug Database (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/). The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 386. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
During treatment of an attack, attention should be given to salt and water balance. For example, if individuals develop hyponatremia, which can induce seizures, they should be treated by saline infusion. Harmful drugs should be stopped. These include barbiturates, sulfonamides, and many others (see the full list here: https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/).
In some patients, an attack is precipitated by a low intake of carbohydrates in an attempt to lose weight. Consequently, dietary counseling is very important. Affected individuals who are prone to attacks should eat a normal balanced diet and should not greatly restrict their intake of carbohydrates or calories, even for short periods of time. If weight loss is desired, it is advisable to contact a physician and dietician.
Premenstrual attacks often resolve quickly with the onset of menstruation. Hormone manipulation may be effective in preventing such attacks and some affected women have been treated with gonadotropin-releasing hormone analogues to suppress ovulation and prevent frequent cyclic attacks. Some individuals who experience recurrent attacks may benefit from regular hematin infusion. This is sometimes recommended for women with severe symptoms during the time of their menses.
If a proper diagnosis has not been made, acute intermittent porphyria (AIP) can be particularly dangerous, especially if drugs which aggravate the disorder are administered. The prognosis of acute intermittent porphyria (AIP) is usually good if the disorder is recognized before severe nerve damage has occurred and if treatment and preventive measures are begun. Although symptoms usually resolve after an attack, some individuals may develop chronic pain. Nerve damage and associated muscle weakness from a severe attack improves over time, but such improvement may take many months to resolve fully.
Liver transplantation has been used to treat some individuals with acute intermittent porphyria (AIP), specifically individuals with severe disease who have failed to respond to other treatment options. A liver transplant in individuals with acute intermittent porphyria (AIP) is an option of last resort. Affected individuals who experience kidney failure may require a kidney transplant. Some individuals have required a combined kidney and liver transplant.
Wearing a Medic Alert bracelet is advisable for patients who have had attacks. People who are asymptomatic carriers of the genetic trait may choose not to wear a bracelet but should be prepared in any medical encounter to advise their care-givers of medications that are risky in acute intermittent porphyria. It should be remembered that acute intermittent porphyria patients can develop other diseases, and symptoms will not always be due to porphyria.
Treatment of Sporadic Acute Neurovisceral Attacks
Intravenous human hemin is the most effective treatment for sporadic acute neurovisceral attacks (i.e., when an individual has experienced 1 to ≤3 acute porphyria attacks). Intravenous administration of hemin preparations may be lifesaving if employed early, when neuronal damage is still reversible, and may help to avoid peripheral neuropathy or prevent its progression.
The recommended dose for hemin is 3-4 mg/kg by IV, given once daily for four days. Treatment may be extended, depending on the clinical course. Note: Because 100 mg of hemin contains 8 mg of iron, frequent administration of hemin may increase the risk for iron overload. Periodic monitoring of serum ferritin concentration and/or transferrin saturation is therefore appropriate in individuals treated repeatedly with hemin.
- Panhematin® is approved for treatment of acute attacks in the United States. This product is supplied as a dried powder, which must be reconstituted with sterile water immediately before IV injection and administered over ten to 15 minutes. Because the administration of Panhematin® reconstituted with sterile water is associated with transient, mild coagulopathy, concurrent anticoagulant therapy should be avoided.
- Heme arginate (Normosang®) is an arginine-stabilized form of human hemin available nearly worldwide, including in Europe, Africa, the Middle East, and South America. It is infused over at least 30 minutes. It has the same advantage as hemin in treating an acute neurovisceral attack but has fewer reported side effects 355.
Note: (1) Phlebitis after IV injection can be minimized by reconstituting hematin in 20% human serum albumin solution and/or by using a large vein or a central catheter for infusion. Peripheral cannulas used to administer hematin should be replaced after each use. (2) An infusion set with an in-line filter is recommended to remove any undissolved particulate matter. (3) Rigorous flushing of venous catheters with boluses of saline totaling 200 mL is recommended.
Recurrent acute attacks are best managed with support and advice from a porphyria specialist. See information and contact details of specialist porphyria centers at the International Porphyria Network website.
If the criterion for recurrent attacks is met, Givlaari® (givosiran) should be considered, as long-term complications of hemin such as iron overload, phlebitis, and loss of venous access can be avoided 361.
Prevention of Recurrent Acute Neurovisceral Attacks
Givlaari® (givosiran) is a subcutaneously delivered RNA interference therapeutic specifically targeting ALAS1 mRNA in the liver to reduce urinary excretion of 5-aminolevulinic acid (ALA) and PBG. It is approved for treatment of acute porphyria in adults and adolescents age ≥12 years in the European Union and adults in the United States. Clinical studies have shown an acceptable safety profile and clinical efficacy in reducing attack rates and use of hemin 367.
When available, use of this treatment has meant that older treatment alternatives such as ovulation suppression therapy and preventive hemin can be avoided. For the sporadic acute attack, hemin is still the treatment of primary choice.
Liver Transplantation
Liver transplantation is curative and reported from several centers 387. Indications include repeated life-threatening acute porphyria attacks and poor quality of life where givosiran is not available or has shown insufficient medical efficacy.
Note that ALA toxicity is the major hypothesis proposed for the pathogenesis of the neurologic lesions causing the clinical features of acute porphyria attacks. Support for this hypothesis are (1) the success of liver transplantation as a cure for recurrent acute attacks 387; and (2) the occurrence of acute attacks in persons who do not have acute intermittent porphyria (AIP) who have received a liver transplant from persons who experience recurrent acute attacks, implicating release of a hepatic neurotoxin, probably ALA, as their cause 388.
Supportive Therapy
Other recommendations to reduce the frequency and/or severity of acute attacks include the following:
- Ensure that adequate nutrition is provided by a normal balanced diet. Avoid unsupervised diets that restrict caloric intake, particularly those that exclude carbohydrates completely.
Seek timely treatment of systemic illness or infection. - Pain relief. Effective analgesia should be provided as soon as possible, usually in the form of parenteral opiates (morphine, diamorphine, and fentanyl are safe). Very large quantities may be required in a severe acute attack. Consider patient-controlled analgesia and support from a pain management team.
- Treatment of hypertension. Sixty percent of symptomatic individuals with acute intermittent porphyria (AIP) have hypertension because of development of acute intermittent porphyria (AIP)-related chronic kidney disease 389. Beta-blockers and renin-angiotensin-aldosterone system (RAAS)-blockers are considered safe and should be used to delay the development of end-stage kidney disease.
- Prevention of nausea and vomiting. Prochloperazine, promazine, or ondansetron are considered safe.
- Prompt treatment of seizures can be terminated with intravenous diazepam, clonazepam, or magnesium sulphate.
- Maintenance of fluid and electrolyte balance. Intravenous fluid replacement may be required to correct dehydration or electrolyte imbalance. Dextrose in water solutions should be avoided because of the risk of hyponatremia. Chronic hyponatremia (developing over >48 hours) should be corrected slowly to minimize the risk of central pontine myelinolysis 380.
- Combined liver and kidney transplantation, which has been successful, can be considered in individuals with acute intermittent porphyria (AIP) who have recurrent acute porphyria attacks and end-stage kidney disease 390. Individuals with acute intermittent porphyria (AIP) may require kidney replacement therapy 362.
Alternative Medical Therapies
Alternative medical therapies to reduce the frequency and/or severity of acute porphyria attacks if givosiran is not available include ovulation suppression therapy and prophylactic hemin infusion.
- Ovulation suppression therapy with gonadorelin analogs has been used for women with recurrent menstrual cycle-related acute neurovisceral attacks 391. Long-acting analogs that can be used to prevent ovulation should be administered during the first few days of the menstrual cycle to minimize the early stimulation effect on hormone release that can trigger an attack. Side effects can be minimized by administering estrogen, preferably by patch. Gynecologic review and bone density monitoring are recommended. This treatment should be continuously evaluated in consultation with a porphyria specialist and preferably not last longer than two years.
- Prophylactic hemin infusion is possible. The minimum effective infusion frequency should be employed, usually a weekly dose of hemin infused via an indwelling venous catheter. Problems include those associated with a venous access device (infection, blockage) and iron overload.
Experimental Therapies
- Enzyme Replacement Therapy [ERT] – Based on the experience of administering doses of recombinant human HMBS/PBGD (rhPBGD) protein in a mouse model of acute intermittent porphyria (AIP) that reduced plasma porphobilinogen (PBG) accumulation during an acute attack induced after phenobarbital challenge, in 2002 the European Medicines Agency (EMA) granted recombinant human HMBS/PBGD an orphan designation (EU/3/ 02/103). Researchers conducted clinical trials in healthy subjects, asymptomatic hydroxymethylbilane synthase (HMBS)-deficient subjects with increased porphyrin precursor excretion, and acute intermittent porphyria (AIP) patients with repeated attacks 392, 393. Although the enzyme was able to detoxify porphobilinogen (PBG) metabolites, the treatment approach limitations included its short half-life in circulation and the lack of liver targeting.
- Liver Gene Therapy – Clinical trials using two strategies, HMBS-gene therapy and interference RNA for ALAS1 gene inhibition, are being conducted in patients with acute intermittent porphyria (AIP). The two strategies include – the delivery of the HMBS gene to the liver cells using a viral vector. The other option is a small interfering RNA (siRNA) directed against aminolevulinic acid synthase, with the objective of reducing delta aminolevulinic acid (ALA) production. Both of them are still in the trial phase and await approval, pending larger trials that would hopefully provide consistent efficacy and safety 49, 394.
Acute intermittent porphyria diet
Acute intermittent porphyria patients prone to attacks should eat a normal or high carbohydrate diet and should not greatly restrict their intakes of carbohydrate and calories, even for short periods of time. If weight loss is desired, it is advisable to consult a physician, who may request that a dietitian estimate an individual’s normal caloric intake, which varies greatly from one person to another. It may be appropriate to prescribe a diet that is approximately 10% below the normal level of calories for the patient. This should result in a gradual weight loss and usually will not cause an attack of porphyria.
Acute intermittent porphyria and pregnancy
Pregnancy in women with acute intermittent porphyria (AIP) is usually uncomplicated. Offspring have a 50% chance of inheriting the gene for acute intermittent porphyria, but the great majority of them remain “latent” for all or most of their lifetimes. The minority that eventually have symptoms usually benefit from treatment. Given these considerations, most patients or individuals with “latent” porphyria elect to have children for the same reasons as anyone else.
Although urinary porphobilinogen (PBG) concentration may increase during pregnancy, this does not lead to a higher frequency of clinical manifestations of porphyria 363.
Preconception counseling is recommended to advise women with acute intermittent porphyria (AIP) of the clinical manifestations of porphyria, self-care, and preventative measures to avoid exacerbations (i.e., adequate and regular nutrition, rest, and carbohydrate intake for treating mild-to-moderate symptoms).
There is a higher risk for pregnancy-induced hypertensive disorder, gestational diabetes, and fetuses with intrauterine growth restriction (IUGR). In general, risk ratios are higher among women with acute intermittent porphyria (AIP) who have high lifetime urinary porphobilinogen (PBG) concentrations 364.
Testing for urinary porphobilinogen (PBG) concentration prior to pregnancy may establish the individual’s risk levels.
Women with biochemically active acute hepatic porphyria (AHP) (i.e., urinary porphobilinogen (PBG) concentration greater than four times the upper limit of normal) or a history of active acute hepatic porphyria (AHP) should be offered specialized prenatal care. Hyperemesis, a catabolic risk for precipitating acute attacks, should be treated promptly. Blood pressure should be monitored once monthly during the first and second trimesters, and weekly during the last trimester. Additional monitoring of fetal growth during pregnancy will help identify intrauterine growth restriction (IUGR) 364.
When a woman with acute intermittent porphyria (AIP) experiences abdominal pain, hypertension, and tachycardia during pregnancy, urine porphobilinogen (PBG) concentration should be measured, and complications of pregnancy should be excluded in consultation with an obstetrician before the findings are attributed to an acute attack.
- If an acute porphyria attack is suspected, a urine porphobilinogen (PBG) concentration should be measured before deciding on specific treatment.
- Any symptomatic treatment needed should be chosen after considering the risk of the drug triggering/aggravating an acute porphyria attack in the pregnant woman.
- An obstetrician should be consulted regarding medical treatment and possible effects on the fetus.
- Human hemin is safe to be used during pregnancy 395
- No human pregnancies have been reported during or after treatment with Givlaari® (givosiran); there are no data on the presence of givosiran in human milk.
Note: In an obstetric emergency, no drug should be restricted if it is likely to be of major clinical benefit or is required in a life-threatening situation.
Acute intermittent porphyria prognosis
Prognosis is good if acute intermittent porphyria is recognized early and treated at the time 329. The mortality rate for acute intermittent porphyria has decreased through the past decades to 5% to 20% (on acute attacks) thanks to new methods of diagnosis and treatment (use of human hemin) 329. But if acute intermittent porphyria is resistant to heme therapy and is recurrent, the only currently approved way to reduce mortality is with an orthotopic liver transplant where a diseased liver is removed and replaced with a healthy donor liver in the same anatomical position 362, 396, 377. The overall survival at five years after liver transplantation is 82%, which is consistent with survival data for individuals transplanted for other metabolic diseases 387.
With ongoing trials exploring the efficacy and safety of enzyme replacement therapy (ERT) and liver gene therapy, the prognosis of acute intermittent porphyria (AIP) is expected to become better very soon 329.
Untreated acute intermittent porphyria (AIP) is associated with significant morbidity and can lead to paresis (incomplete paralysis) and death. Long-term complications include chronic hypertension, chronic neuropathy, chronic kidney disease, and risk of hepatocellular carcinoma (liver cancer) 397, 398, 367.
Variegate porphyria
Variegate porphyria is caused by a mutation in the enzyme protoporphyrinogen oxidase, which is part of the pathway that produces porphyrins and heme. Acute attacks are similar to those in Acute intermittent porphyria and hereditary coproporphyria but are unusual. A more common sign of the disease is blistering skin lesions, which are chronic in many people with variegate porphyria.
Acute attacks almost always start with severe pain in the abdomen but sometimes in the chest, back, or thighs, and are often accompanied by nausea, vomiting, and constipation. Heart rate and blood pressure are commonly increased. These symptoms and signs are all due to the effects of the disease on the nervous system. Confusion, convulsions, and muscular weakness, due to impairment of the nerves controlling the muscles, may lead to paralysis. An acute attack usually lasts for days or weeks. Recovery from severe paralysis is generally slow.
Variegate porphyria is especially common in South Africa in individuals of Dutch ancestry, where it has been estimated that 3 in 1,000 of the white population are affected. It is much less prevalent in other countries. Like acute intermittent porphyria and hereditary coproporphyria, it is an autosomal dominant disorder, meaning that a mutation is present in only one of the pair of protoporphyrinogen oxidase genes.
Figure 29. Variegate porphyria autosomal dominant inheritance pattern
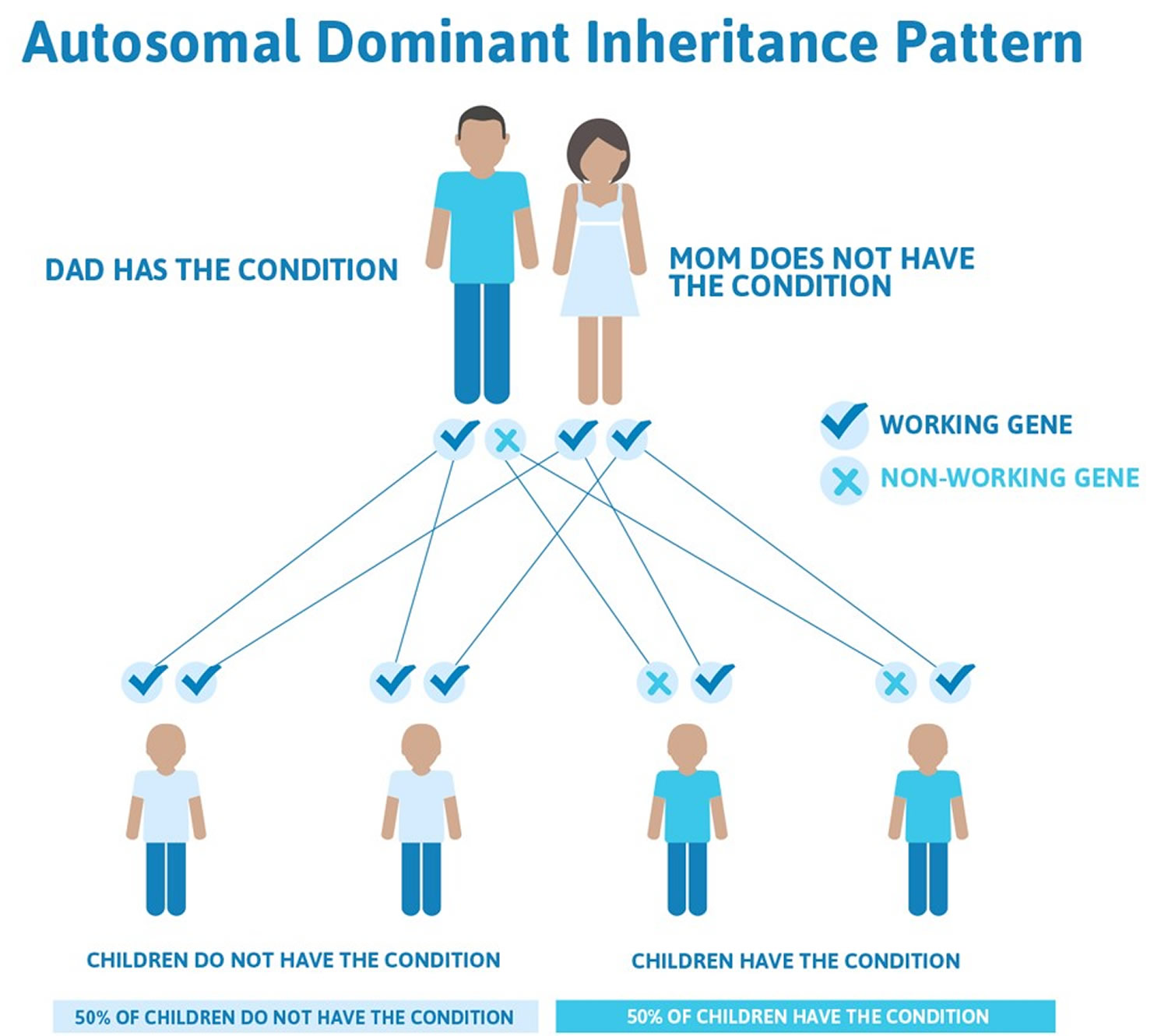
Variegate porphyria causes
As in hereditary coproporphyria, acute attacks of variegate porphyria are unusual except in the presence of environmental activating factors, such as drugs, hormones, and dietary changes.
Variegate porphyria is caused by mutations of the protoporphyrinogen oxidase gene. A protoporphyrinogen oxidase mutation is inherited as an autosomal dominant trait within a family. The pattern of inheritance is autosomal dominant, which means that a single mutation is inherited from one parent and, in the presence of other triggering factors, is sufficient to cause the disease. The abnormal gene can be inherited from either parent, or on rare occasions can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50 percent for each pregnancy regardless of the sex of the resulting child.
The protoporphyrinogen oxidase gene is located on the long arm (q) of chromosome 1 (1q22). Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 1q22” refers to band 22 on the long arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
The protoporphyrinogen oxidase gene contains instructions for creating protoporphyrinogen oxidase, one of the eight enzymes necessary for the production of heme. Heme is an iron-containing porphyrin (iron protoporphyrin) and is a part of many heme-containing proteins (hemoproteins) in the body. Hemoproteins interact with oxygen and some are involved in electron transport and energy metabolism. The best known hemoprotein is hemoglobin, which is made in the bone marrow, makes red blood cells red, and transports oxygen from the lungs to other tissues. However, the bone marrow and hemoglobin are not affected in variegate porphyria. In this condition the heme pathway in the liver, which makes heme for other important hemoproteins, is affected.
Mutations of the protoporphyrinogen oxidase gene result in deficient levels of protoporphyrinogen oxidase, which, in turn, disrupts the biochemical process to create heme in the liver. This disruption causes porphyrins and porphyrin precursors to accumulate in the liver and these are then transported to other parts of the body to affect the nervous system and skin.
A variety of different triggers are known to lead to attacks in individuals with variegate porphyria. Many of these triggers act by increasing heme synthesis in the liver, which makes the protoporphyrinogen oxidase deficiency more significant and increases the accumulation of porphyrins and porphyrin precursors. As noted above, triggers include a variety of drugs, hormones (especially progesterone), reduced intake of calories and carbohydrate, alcohol, and stress induced by infection or other illness.
Variegate porphyria diagnosis
Urine aminolevulinic acid and porphobilinogen are increased during attacks, but as in hereditary coproporphyria, these may increase less and decrease more rapidly than in acute intermittent porphyria. Plasma porphyrins are frequently increased in variegate porphyria, in contrast to acute intermittent porphyria and hereditary coproporphyria, and the plasma of variegate porphyria patients displays a distinctive fluorescence peak, which is diagnostic. Fecal porphyrins are also elevated and are predominantly coproporphyrin III and protoporphyrin.
Molecular genetic testing to identify a protoporphyrinogen oxidase mutation is recommended for all biochemically confirmed cases of variegate porphyria. Molecular testing is sometimes useful when symptoms have been absent for months or years and biochemical abnormalities are no longer present. Knowing the protoporphyrinogen oxidase mutation is a family enables other family members to be tested reliably for the same mutation.
Variegate porphyria treatments
Management and prevention are the same as in acute intermittent porphyria and hereditary coproporphyria. Hospitalization is usually indicated for pain control and treatment of other severe symptoms such as nausea and vomiting, electrolyte imbalances and convulsions. Monitoring for these manifestations as well and muscle weakness and respiratory embarrassment is also indicated in severe attacks. A narcotic analgesic is generally required for pain, and a phenothiazine or ondansetron for nausea and vomiting. Triggering factors should be identified and discontinued when possible. Specific therapies are hemin for injection, which is available in the U.S. as lyophilized hematin (Panhematin®,), and glucose loading. Hemin represses the heme pathway in the liver and lowers aminolevulinic acid, porphobilinogen and porphyrins, and is associated with more rapid recovery from an attack. Glucose given intravenously has a similar effect, but because it is less potent is used only for mild attacks, or until hemin can be obtained from the manufacturer. Blistering skin lesions are much more common than in hereditary coproporphyria and are not readily treated. The only effective preventive measure is use of protective clothing.
Variegate porphyria prognosis
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic Variegate Porphyria carriers should become informed on medications to avoid and should be prepared to point their healthcare providers to on-line drug lists that are regularly updated.
A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue (Lupron, Zoladex, others) administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite on-line discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction (ca. 10%), which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
Hereditary coproporphyria
Hereditary coproporphyria (HCP) is a rare inherited form of liver (hepatic) porphyria, that involve defects in heme or ‘haem’ biosynthetic pathway caused by mutations (changes) in the CPOX gene which results in deficiency of the enzyme coproporphyrinogen oxidase or coproporphyrinogen-III oxidase (CPOX), the sixth enzyme in the heme biosynthetic pathway that produces porphyrins and heme. Hereditary coproporphyria (HCP) is characterized by neurological symptoms in the form of episodes (acute attacks) of stomach pain, nausea, vomiting, weakness, numbness, and pain in the hands and feet (neuropathy) 399, 34, 400, 401, 402, 403, 404, 405, 406, 407. The porphyrias are a group of blood conditions caused by a lack of an enzyme in the body that makes heme or haem, an important molecule that carries oxygen throughout the body and is vital for all of the body’s organs 408.
Hereditary coproporphyria (HCP) is caused by mutations in the CPOX (coproporphyrinogen oxidase) gene that provides instructions for making an enzyme known as coproporphyrinogen oxidase (CPOX). The enzyme coproporphyrinogen oxidase or coproporphyrinogen-III oxidase (CPOX) enzyme is involved in the production of a molecule called heme (haem). Heme is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme or haem is an essential component of iron (Fe) containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood), which is necessary to bind oxygen in the bloodstream and transport of oxygen in your body. Heme molecule also helps in respiration, detoxification of drugs, and other different biological functions. In hemoglobin (Hb), iron exists as ferrous (Fe2+) iron ion that is located at the center of the porphyrin ring, held in place by the four nitrogen atoms of the pyrrole rings. Normally, your body makes heme (haem) in a multi-step process that requires 8 different enzymes. Porphyrins are made during several steps of this process. Coproporphyrinogen oxidase (CPOX) enzyme is responsible for the sixth step in this process, the removal of carbon and oxygen atoms from coproporphyrinogen III (the product of the fifth step) to form protoporphyrinogen IX (PPIX). In subsequent steps, two other enzymes modify protoporphyrinogen IX (PPIX) and incorporate an iron atom to produce heme.
In people with hereditary coproporphyria (HCP), the coproporphyrinogen oxidase (CPOX) enzyme deficiency results in the accumulation of porphyrin precursors in their body. Symptoms usually begin around 20 to 30 years of age, but have been reported at younger ages. Signs and symptoms present during the attacks may include body pain, nausea and vomiting, increased heart rate (tachycardia) and high blood pressure 399. Less common symptoms include seizures, skin lesions, and paralysis of the arms and legs, body trunk, and respiratory muscles. Most individuals with hereditary coproporphyria do not have any signs or symptoms between attacks 409.
In hereditary coproporphyria (HCP) the CPOX gene mutation is inherited in an autosomal dominant manner meaning that a mutation is present in only one of the pair of coproporphyinogen oxidase (CPOX) genes. However, the enzyme coproporphyrinogen oxidase (CPOX) deficiency by itself is not sufficient to produce symptoms of the disease and most individuals with a CPOX gene mutation do not develop symptoms of hereditary coproporphyria (HCP). Additional factors such as endocrine factors (e.g. hormonal changes), the use of certain drugs, excess alcohol consumption, infections, and fasting or dietary changes are required to trigger the appearance of symptoms 408. Some affected individuals experience acute attacks or episodes that develop over a period of days. The course and severity of attacks is highly variable from one person to another. In some cases, particularly those without proper diagnosis and treatment, the disorder can cause life-threatening complications.
Hereditary coproporphyria (HCP) diagnosis is based on the symptoms and specific blood, urine and stool testing. Treatment is based on preventing the symptoms. An acute attack requires hospitalization, medications, and treatment with heme therapy 410.
Figure 30. Hereditary coproporphyria (HCP)
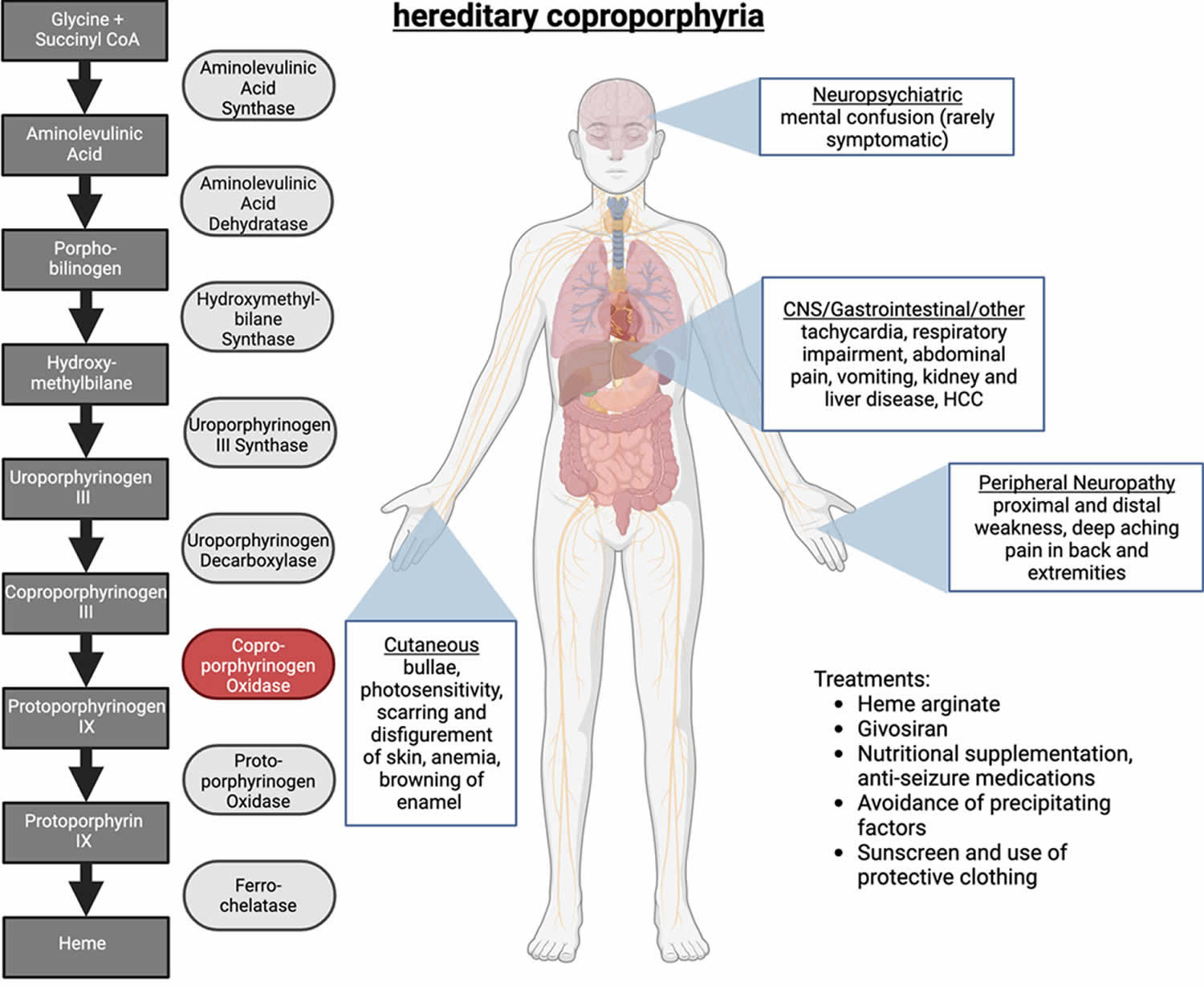
Footnotes: In hereditary coproporphyria (HCP), coproporphyrinogen and uroporphyrinogen accumulate in patients, causing acute attacks, abdominal pain, and motor neuropathy. Skin signs and symptoms including photosensitivity also occur. Heme arginate is an effective treatment for hereditary coproporphyria (HCP), and Givosiran may also prevent the development of symptoms.
[Source 34 ]Hereditary coproporphyria diagnosis
The initial test for people with symptoms is quantitative urinary aminolevulinic acid, porphobilinogen and porphyrins. Elevation of aminolevulinic acid, porphobilinogen and coproporphyrin (predominantly isomer III) is highly suggestive of Hereditary coproporphyria. For asymptomatic individuals, the urine studies may be normal, but a fecal porphyrin analysis will show elevation of coproporphyrin III. Screening tests of this kind should be confirmed by DNA analysis to confirm a coproporphyinogen oxidase mutation.
Hereditary coproporphyria treatment
There is no specific treatment for hereditary coproporphyria (HCP). Treatment is aimed at managing the symptoms of hereditary coproporphyria that occur during an acute attack. Hospitalization is often necessary for acute attacks, and medications for pain, nausea and vomiting, and close observation are generally required. Initial treatment steps include stopping any medications that can potentially worsen hereditary coproporphyria or cause an attack. All triggering factors should be identified, if possible, and discontinued. In addition, ensuring proper intake of carbohydrate, either orally or intravenously, is essential. Wearing a Medic Alert bracelet or the use of a wallet card is advisable in individuals who have hereditary coproporphyria.
Some attacks may be managed by giving the person a large amount of glucose or other carbohydrates. More severe attacks are treated with heme therapy, given through a vein (intravenously). Panhematin® (hemin for injection), an enzyme inhibitor derived from red blood cells that is potent in suppressing acute attacks of porphyria, is currently the only commercially available heme therapy for treatment and prevention of acute porphyria attacks in the United States. Heme arginate (Normosang®), which is marketed in some other countries, is another type of heme therapy 410. Panhematin® (hemin for injection) almost always returns porphyrin and porphyrin precursor levels to normal values. The U.S. Food and Drug Administration (FDA) originally approved panhematin for the treatment of recurrent attacks of acute intermittent porphyria (AIP) related to the menstrual cycle in susceptible women. Numerous symptoms including pain, hypertension, tachycardia and altered mental status, and neurologic signs have improved in individuals with acute porphyria after treatment with panhematin. Because of its potency, it is usually given after a trial of high-dose carbohydrate of any sort, including glucose therapy and should be administered only by physicians experienced in the management of porphyrias in a hospital setting. Responses to hematin infusion can include reduced urine concentration of porphobilinogen (PBG) (the 1st sign) after 2 doses; clinical improvement after 3 to 4 doses (typically dramatic) with no further need of narcotic analgesia 399. Hemin treatment withdrawal triggered a rapid return of clinical symptoms 411, 412. Furthermore, heme arginate (Normosang®) has been demonstrated as an effective maintenance therapy for patients with hereditary coproporphyria (HCP), and can significantly improve quality of life 399, 411.
Some individuals who experience recurrent attacks may benefit from chronic hematin infusion. This is sometimes recommended for women with severe symptoms during the time of their menses.
Attacks can also be avoided by avoiding the ‘trigger’ for the attack. The trigger can be different for different people. Triggers include certain medications, alcohol, dieting, and hormonal changes in the body. Sometimes, however, the trigger is unknown. If the person with hereditary coproporphyria has skin symptoms, avoiding excess sun exposure can reduce the blisters and skin lesions 413.
In 2019, the FDA approved Givosiran (Givlaari) to treat adults with acute hepatic porphyria, including hereditary coproporphyria (HCP). Givosiran (Givlaari) is a small interfering RNA (siRNA) that targets the mRNA of the enzyme aminolevulinate synthase 1 (ALAS1). By doing so, it reduces the production of aminolevulinic acid (ALA) and porphobilinogen (PBG), the toxic substances that accumulate in acute hepatic porphyria. Givlaari aims to prevent attacks from occuring and may reduce or prevent the development of neurologic symptoms in persons with hereditary coproporphyria (HCP); however, this remains to be determined with long-term studies 399. In a phase 3 trial of Givosiran, only one individual with hereditary coproporphyria (HCP) was included in the study, although reports indicate clinical response 414.
Other treatments include glucose administration to reverse fasting state, dextrose rehydration for hyponatremia, and anti-seizure medications 399.
Treatment for hereditary coproporphyria may also include drugs to treat specific symptoms such as certain pain medications (analgesics), anti-anxiety drugs, anti-hypertensive drugs, and drugs to treat nausea and vomiting, tachycardia, or restlessness. Medications to treat any infections that may occur at the same time as an attack (intercurrent infection) may also be necessary. Seizures may require treatment with anti-seizure (anti-convulsant) medications, but many of the common options can worsen an attack and are contraindicated. A short-acting benzodiazepine or magnesium may be recommended. Gabapentin and propofol are considered effective and safe for prolonged control of seizures.
Although many types of drugs are believed to be safe in individuals with hereditary coproporphyria, recommendations about drugs for treating hereditary coproporphyria are based upon experience and clinical study. Since many commonly used drugs have not been tested for their effects on porphyria, they should be avoided if at all possible. If a question of drug safety arises, a physician or medical center specializing in porphyria should be contacted. The American Porphyria Foundation offers a drug database (https://porphyriafoundation.org/drugdatabase/drug-safety-database-search) with safety information about the interaction of specific drugs in patients with porphyria.
Additional treatment for individuals undergoing an attack includes monitoring for muscle weakness and respiratory issues and monitoring fluid and electrolyte balances. For example, if affected individuals develop low blood sodium level (hyponatremia), which can induce seizures, they should be treated by restricting the intake of water (water deprivation). If serum sodium is decreased severely, e.g. from normal (>134 meq/dl) to very low (100-115 meq/dl), then saline infusion is indicated.
Premenstrual attacks often resolve quickly with the onset of menstruation. Hormone manipulation may be effective in preventing such attacks. Some affected women have been treated with gonadotropin-releasing hormone analogues to suppress ovulation and prevent frequent cyclic attacks.
In some cases, an attack is precipitated by a low intake of carbohydrates in an attempt to lose weight. Consequently, dietary counseling is very important. Affected individuals who are prone to attacks should eat a normal carbohydrate diet and should not greatly restrict their intake of carbohydrates or calories, even for short periods of time. If weight loss is desired, it is advisable to contact a physician and dietitian.
In individuals who develop skin complications, avoidance of sunlight will be of benefit and can include the use of double layers of clothing, long sleeves, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective. Affected individuals will also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars. Avoidance of sunlight can potentially cause vitamin D deficiency and some individuals may require supplemental vitamin D.
A liver transplant has been used to treat some individuals with acute forms of porphyria, specifically individuals with severe disease who have failed to respond to other treatment options. A liver transplant in individuals with hereditary coproporphyria is an option of last resort.
Hereditary coproporphyria prognosis
The prognosis is usually good if the disease is recognized and treated promptly, before nerve damage develops. Although symptoms usually resolve after an attack, recovery of neuromuscular function (in a severe case) may require several months or longer. Mental symptoms may occur during attacks but are not chronic. Premenstrual attacks often resolve quickly with the onset of menses.
Can attacks be prevented?
Yes, particularly with regard to drugs and diet. Genetic hereditary coproporphyria carriers should become informed on drugs and other factors that can lead to symptoms (see above). They should be prepared to point their healthcare providers to drugs and medications to avoid. A Medic Alert bracelet is useful for a situation in which the patient is incapacitated. Very frequent premenstrual attacks can be prevented by a gonadotropin-releasing hormone (GnRH) analogue administered with expert guidance. In selected cases, frequent noncyclic attacks can be prevented by once- or twice-weekly infusions of hemin.
Individuals who are prone to attacks should consume a normal balanced diet. Despite online discussion, there is no evidence that pushing carbohydrate prevents attacks, and it has the side effect of weight gain, which is undesirable for most people. Fasting, fad diets (for example, high protein) and gastric reduction surgery should be avoided. If weight loss is desired, it is advisable to consult a physician and a dietitian about an individualized diet with modest caloric restriction, which will produce gradual weight loss without increasing the risk of an attack of porphyria. Exercise is safe in porphyria, and recommended.
ALA dehydratase deficiency porphyria
ALA dehydratase deficiency porphyria is a severe disorder caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase which results in an increase of 5’-aminolevulinic acid (ALA) in the liver, other tissues, blood plasma, and urine. In addition, urine coproporphyrin and erythrocyte protoporphyrin are increased. ALA dehydratase deficiency porphyria generally presents with sudden attacks of severe stomach pain that last for several days.
All of the reported cases of ALA dehydratase deficiency porphyria have been males, in contrast to the other acute porphyrias. ALA dehydratase deficiency porphyria is the least common of all the porphyrias with less than 10 cases documented to date. This is an autosomal recessive disease, whereas the other three acute porphyrias are autosomal dominant. Each parent of an affected individual must have a mutation in one of their δ-aminolevulinic acid dehydratase genes and both must pass their mutation on to their child.
ALA-dehydratase porphyria causes
ALA dehydratase deficiency porphyria is caused by a deficiency of the enzyme δ-aminolevulinic acid dehydratase (ALAD).
ALA dehydratase deficiency porphyria is caused by mutations in the ALA dehydratase deficiency gene, and the disease is inherited as an autosomal recessive disorder. This means that both copies of the ALA dehydratase deficiency gene have a mutation. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother.
Recessive genetic disorders occur when an individual inherits two copies of an abnormal gene for the same trait, one from each parent. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females. Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
The ALA dehydratase deficiency gene contains instructions for creating the enzyme aminolevulinate dehydratase, which is necessary for the production of heme. Heme is part of hemoglobin, which is the oxygen-carrying component of red blood cells. Heme is mainly produced in the bone marrow and the liver. Eight different enzymes are necessary for the creation of heme.
Mutations of the ALA dehydratase deficiency gene result in deficient levels ofporphobilinogen in the body, with accumulation of ALA, which causes the symptoms associated with ALA dehydratase deficiency porphyria.
A variety of different triggers have been identified that can precipitatean acute attack in individuals with ALA dehydratase deficiency porphyria. These triggers include alcohol, certain drugs, physical and psychological stress, infection, fasting (reduced caloric intake) and dehydration. The use of estrogen or progesterone is also suspect of triggering an acute attack.
ALA dehydratase deficiency porphyria diagnosis
There are many laboratory tests available for the porphyrias, and it is often difficult to decide which should be chosen. Many of these tests are expensive and the results are often difficult to interpret. When abdominal and neurological symptoms suggest an acute porphyria, the best screening tests are urinary aminolevulinic acid (ALA) and porphobilinogen (PBG). DNA testing to identify the specific mutation in an individual’s porphyria-causing gene is the most specific and sensitive test to confirm the diagnosis of a specific porphyria. Before requesting DNA testing, it is recommended that patients have biochemical testing (urinary, stool and/or plasma porphyrins and porphyrin precursors (ALA and PBG) and/or enzyme assays). However, biochemical testing may be inconclusive.
ALA dehydratase deficiency porphyria treatment
Treatment is the same as in the other acute porphyrias. For the acute porphyrias, hospitalization is often necessary for acute attacks. Medications for pain, nausea and vomiting, and close observation are generally required with monitoring of salt and water balance. Harmful drugs should be stopped. Attacks are treated with either glucose loading or intravenous administration of hemin (Panhematin®). Attacks can be prevented in many cases by avoiding harmful drugs and adverse dietary practices.
Porphyria complications
Possible complications depend on the form of porphyria:
- Acute porphyrias can be life-threatening if an attack isn’t promptly treated. During an attack, you may experience dehydration, breathing problems, seizures and high blood pressure. Episodes often require hospitalization for treatment. Long-term complications with recurrent acute attacks may include chronic pain, chronic kidney failure and liver damage.
- Cutaneous porphyrias can result in permanent skin damage. Also, the skin blisters can become infected. When your skin heals after cutaneous porphyria, it may have an abnormal appearance and coloring, be fragile, or leave scars.
Without medical treatment, complications of porphyria may include:
- Permanent hair loss
- Skin scarring
- Permanent skin pigmentation changes
- Dehydration
- Breathing problems
- High blood pressure (hypertension)
- Low salt levels in the blood (hyponatremia)
- Kidney failure
- Liver problems, which may require a liver transplant in severe cases. Several types of porphyrias can cause liver problems. Acute porphyria increases the chance of developing liver cancer (hepatocellular carcinoma [HCC]). Porphyria cutanea tarda can damage the liver and increase the chance of developing cirrhosis (scarring of the liver) and liver cancer. Some people with protoporphyria also develop liver damage and cirrhosis, and up to 5 percent of people with protoporphyria develop liver failure 415. In people with protoporphyria, bile carries extra porphyrins from the liver to the gallbladder, which may lead to gallstones that are made of porphyrins.
- Anemia. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria who develop severe anemia may require treatment with blood transfusions. In some cases, doctors may also recommend surgery to remove an enlarged spleen, which can help treat anemia.
Porphyria causes
The substance heme (or haem) is used in various metabolic processes. The body makes heme from porphyrins, which are metallic compounds found naturally in the tissues of animals and plants. The conversion of porphyrins into heme requires the action of special proteins called enzymes. Genes control the action of enzymes. A flawed gene (or genes) can stop the body from making one or more of these enzymes. This creates a lack of heme and a build-up of porphyrins, which causes the signs and symptoms of porphyria.
Genetic forms
Most forms of porphyria are inherited, which means the genetic predisposition is passed from one generation to the next. The faulty gene interferes with the body’s ability to create one or more enzymes necessary in the conversion of porphyrins into heme. The pattern of inheritance may include:
- Autosomal dominant inheritance – the faulty gene is inherited from one parent. This faulty gene overrides the healthy gene inherited from the other parent.
- Autosomal recessive inheritance – the faulty gene is inherited from both parents.
However, about nine in every 10 people with the faulty gene or genes don’t have porphyria. It appears that an environmental trigger is needed to allow porphyria to develop. You might have what’s called latent porphyria, and never have symptoms. This is the case for most carriers of the abnormal genes.
Acquired forms
Porphyria cutanea tarda typically is acquired rather than inherited, although the enzyme deficiency may be inherited. Certain triggers that impact enzyme production — such as too much iron in the body, liver disease, estrogen medication, smoking or excessive alcohol use — can cause symptoms.
Porphyria cutanea tarda is usually an acquired disorder, meaning factors other than genes cause the enzyme deficiency. This type of porphyria can be triggered by
- too much iron
- use of alcohol or estrogen
- smoking
- chronic hepatitis C—a long-lasting liver disease that causes inflammation, or swelling, of the liver
- HIV—the virus that causes AIDS
- abnormal genes associated with hemochromatosis—the most common form of iron overload disease, which causes the body to absorb too much iron
Risk factors for porphyria
In addition to genetic risks, environmental factors may trigger the development of signs and symptoms in porphyria. When exposed to the trigger, your body’s demand for heme production increases. This overwhelms the deficient enzyme, setting in motion a process that causes a buildup of porphyrins.
Examples of triggers include:
- Exposure to sunlight
- Certain medications, including hormone drugs
- Recreational drugs
- Dieting or fasting
- Smoking
- Physical stress, such as infections or other illnesses
- Emotional stress
- Alcohol use
- Menstrual hormones ― acute porphyria attacks are rare before puberty and after menopause in women.
Porphyria symptoms
The signs and symptoms of porphyria can vary, depending on the type and severity. Some people have no symptoms. Some go for long periods without any symptoms. Some people have quite a bit of trouble with symptoms.
The most common symptoms are:
- skin problems like sensitivity to the sun, blistering, discoloration and scarring
- abdominal pain
- muscle weakness
- numbness in the arms and legs
- confusion and seizures.
Symptoms vary from one type of porphyria to the next. Cases are generally classified into one of three groups, which include:
- Acute porphyrias – the condition mostly affects the nervous system. The skin is occasionally affected. Symptoms may include muscle pain or paralysis, seizures, disorientation, hallucination, bloody (red) urine, hypertension and gastrointestinal problems such as vomiting, abdominal pain and constipation. Acute porphyrias generally occur during adulthood and are rare before puberty or after menopause. Different types of acute porphyria include ‘acute intermittent porphyria’ and ‘erythropoietic protoporphyria’.
- Signs and symptoms of acute porphyria may include:
- Severe abdominal pain
- Pain in your chest, legs or back
- Constipation or diarrhea
- Nausea and vomiting
- Muscle pain, tingling, numbness, weakness or paralysis
- Red or brown urine
- Mental changes, such as anxiety, confusion, hallucinations, disorientation or paranoia
- Breathing problems
- Urination problems
- Rapid or irregular heartbeats you can feel (palpitations)
- High blood pressure
- Seizures
- Signs and symptoms of acute porphyria may include:
- Cutaneous porphyrias – the condition affects the skin but not the nervous system. The skin is highly sensitive to sunlight and exposure tends to trigger symptoms within minutes. Symptoms may include red, itchy, blistered, painful and swollen skin and bloody (red) urine. The condition may develop during childhood. Different types of cutaneous porphyria include ‘porphyria cutanea tarda’ and ‘hepatoerythropoietic porphyria’.
- As a result of sun exposure, you may experience:
- Sensitivity to the sun and sometimes artificial light, causing burning pain
- Sudden painful skin redness (erythema) and swelling (edema
- Blisters on exposed skin, usually the hands, arms and face
- Fragile thin skin with changes in skin color (pigment)
- Itching
- Excessive hair growth in affected areas
- Red or brown urine
- As a result of sun exposure, you may experience:
- Neurocutaneous porphyrias – the condition affects both the skin and the nervous system. Sunlight exposure tends to rapidly trigger symptoms. Different types of neurocutaneous porphyria include ‘variegate porphyria’ and ‘hereditary coproporphyria’.
Porphyria diagnosis
Because porphyria can cause so many different symptoms, it can be hard to diagnose. Your doctor can talk to you and examine you, and will probably want to arrange urine or blood tests as well.
Genetic tests can be useful, too.
Since porphyria is rare, most doctors are unfamiliar with it and may not recognise the symptoms. Diagnosis can be delayed because porphyria mimics the symptoms and signs of various other medical conditions such as Guillain-Barre syndrome, eczema, multiple sclerosis and irritable bowel syndrome. Diagnostic tests may include:
- Physical examination
- Medical history
- Urine tests to check for elevated substances including porphyrins
- Blood tests to check for high levels of porphyrins in the plasma
- Stool sample to check for excreted porphyrins
- Genetic test.
Porphyria treatment
Treatment for porphyria will depend on what type of porphyria you have and how severe your symptoms are.
Some of the medicines used to treat a sudden (acute) attack of porphyria may include:
- Hematin given through a vein (intravenously)
- Pain medicine
- Propranolol to control the heartbeat
- Sedatives to help you feel calm and less anxious
Other treatments may include:
- Beta-carotene supplements to lessen photosensitivity
- Chloroquine in low doses to reduce levels of porphyrins
- Fluids and glucose to boost carbohydrate levels, which helps limit the production of porphyrins
- Removal of blood (phlebotomy) to reduce levels of porphyrins
Depending on the type of porphyria you have, your doctor may tell you to:
- Avoid all alcohol
- Avoid certain drugs that may trigger an attack
- Avoid injuring the skin
- Avoid sunlight as much as possible and use sunscreen when outside
- Eat a high-carbohydrate diet
If a question of drug safety arises, a physician or medical center specializing in porphyria should be contacted. A list of these institutions may be obtained from the American Porphyria Foundation. The Foundation also maintains an Acute Porphyria Drug Database (https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/). The EPNET/NAPOS Database should also be consulted. The Norwegian Porphyria Centre (NAPOS), with the European Porphyria Network (EPNET), has created a list of medications that clinicians must avoid using in porphyria patients (https://drugsporphyria.net/). These drugs include ketamine, thiopental, chloramphenicol, erythromycin, nitrofurantoin, rifampicin, trimethoprim/sulfamethoxazole, spironolactone, methyldopa, valproic acid, carbamazepine, phenytoin, phenobarbital, primidone, and risperidone 386. For information on prescribing medication in the context of certain conditions (e.g., HIV, epilepsy, malaria), see https://porphyria.uct.ac.za/porphyria-professionals/prescribing-porphyria-treatment-specific-disorders-poprhyria/therapy-epilepsy.
During treatment of an attack, attention should be given to salt and water balance. For example, if individuals develop hyponatremia, which can induce seizures, they should be treated by saline infusion. Harmful drugs should be stopped. These include barbiturates, sulfonamides, and many others (see the full list here: https://porphyriafoundation.org/for-healthcare-professionals/ahp-drug-safety-database/).
Acute porphyria
Doctors most often treat acute porphyria attacks in a hospital. A doctor treats acute porphyrias with heme or glucose loading to decrease the liver’s production of porphyrins and porphyrin precursors. A patient receives heme intravenously once a day for 4 days. Glucose loading involves giving a patient a glucose solution by mouth or intravenously. Heme is usually more effective and is the treatment of choice unless symptoms are mild. In rare instances, if symptoms are severe, a doctor will recommend liver transplantation to treat acute porphyria. In liver transplantation, a surgeon removes a diseased or an injured liver and replaces it with a healthy, whole liver or a segment of a liver from another person, called a donor. A patient has liver transplantation surgery in a hospital under general anesthesia. Liver transplantation can cure liver failure.
Treatment of acute porphyria attacks focuses on providing rapid treatment of symptoms and preventing complications. Treatment may include:
- Pain medication
- Addressing the underlying cause – for example, prescribing antibiotics to treat an infection or ceasing a particular medication
- Medication called ‘hematin’, which is a type of heme the body can use
- Intravenous fluids and glucose
- Admission to hospital in severe cases.
There are plenty of ways to prevent future porphyria attacks.
To prevent future attacks, your doctor may recommend:
- Avoiding any drugs that may trigger an attack in people who have porphyria. Talk with your doctor before you take any over-the-counter or prescription medicines, dietary supplements or complementary or alternative medicines, such as herbal or botanical medicines.
- Avoiding alcohol. Experts recommend no more than one drink per day for women and no more than two drinks per day for men 416.
- Protecting your skin from the sun as much as possible
- Eating carbohydrates frequently
- Eating a balanced diet and avoiding fasting or extreme diets.
- Avoiding smoking
Avoiding triggers may include:
- Not using medications known to trigger acute attacks. Ask your doctor for a list of safe and unsafe drugs.
- Not using alcohol or recreational drugs.
- Avoiding fasting and dieting that involves severe calorie restriction.
- Not smoking.
- Taking certain hormones to prevent premenstrual attacks.
- Minimizing sun exposure. When you’re outdoors, wear protective clothing, and use an opaque blocking sunscreen, such as one with zinc oxide. When
- indoors, use window filters.
- Treating infections and other illnesses promptly.
- Taking steps to reduce emotional stress.
Your doctor may also advise you about:
- medication to control pain, or any nausea or vomiting
- other medication to reduce the amount of porphyrin in the blood
- regular blood donation or blood-letting to reduce the amount of porphyrin in the blood.
Cutaneous porphyria
Treatment may include:
- Oral administration of activated charcoal, which helps to absorb excess porphyrins
- Daily supplementation with beta-carotene (vitamin A) as part of long-term treatment.
- A dietary supplement to replace vitamin D deficiency caused by avoidance of sunlight.
- Periodically drawing blood (phlebotomy) to reduce the iron in your body, which decreases porphyrins.
- Taking a drug used to treat malaria — hydroxychloroquine (Plaquenil) or, less often, chloroquine (Aralen) — to absorb excess porphyrins and help your body get rid of them more quickly than usual. These medications are generally used only in people who can’t tolerate a phlebotomy.
The most important step a person can take to treat a cutaneous porphyria is to avoid sunlight as much as possible. Other cutaneous porphyrias are treated as follows:
- Porphyria cutanea tarda. A health care provider treats porphyria cutanea tarda by removing factors that tend to activate the disease and by performing repeated therapeutic phlebotomies to reduce iron in the liver. Therapeutic phlebotomy is the removal of about a pint of blood from a vein in the arm. A technician performs the procedure at a blood donation center, such as a hospital, clinic, or bloodmobile. A patient does not require anesthesia. Another treatment approach is low-dose hydroxychloroquine tablets to reduce porphyrins in the liver.
- Erythropoietic protoporphyria. People with erythropoietic protoporphyria may be given beta-carotene or cysteine to improve sunlight tolerance, though these medications do not lower porphyrin levels. Experts recommend hepatitis A and hepatitis B vaccines and avoiding alcohol to prevent protoporphyric liver failure. A health care provider may use liver transplantation or a combination of medications to treat people who develop liver failure. Unfortunately, liver transplantation does not correct the primary defect, which is the continuous overproduction of protoporphyria by bone marrow. Successful bone marrow transplantations may successfully cure erythropoietic protoporphyria. A health care provider only considers bone marrow transplantation if the disease is severe and leading to secondary liver disease.
- Congenital erythropoietic porphyria and hepatoerythropoietic porphyria. People with congenital erythropoietic porphyria or hepatoerythropoietic porphyria may need surgery to remove the spleen or blood transfusions to treat anemia. A surgeon removes the spleen in a hospital, and a patient receives general anesthesia. With a blood transfusion, a patient receives blood through an intravenous (IV) line inserted into a vein. A technician performs the procedure at a blood donation center, and a patient does not need anesthesia.
Eating, Diet, and Nutrition
People with an acute porphyria should eat a diet with an average-to-high level of carbohydrates. The recommended dietary allowance for carbohydrates is 130 g per day for adults and children 1 year of age or older; pregnant and breastfeeding women need higher intakes 416. People should avoid limiting intake of carbohydrates and calories, even for short periods of time, as this type of dieting or fasting can trigger symptoms. People with an acute porphyria who want to lose weight should talk with their doctor and dietitian about diets they can follow to lose weight gradually.
People undergoing therapeutic phlebotomies should drink plenty of milk, water, or juice before and after each procedure.
A health care provider may recommend vitamin and mineral supplements for people with a cutaneous porphyria.
Home remedies
Be guided by your doctor, but general suggestions include:
- In all cases avoid known triggers – for example, don’t smoke.
- When out in the sun, wear sunglasses, a brimmed hat, a long-sleeved top and long pants. Apply SPF 30+ sunscreen to exposed skin areas.
- Protect your skin every day. For example, wear rubber gloves when handling chemicals or very hot water. Avoid perfumed soaps. Regularly apply barrier cream to the hands.
- Eat regular meals and avoid alcohol.
- You may like to consider wearing a medical alert bracelet or pendant, since surgery and some drugs can provoke symptoms.
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. New England Journal of Medicine. 2017;377(9):862–872. doi: 10.1056/NEJMra1608634[↩][↩]
- Balwani M, Bloomer J, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Erythropoietic Protoporphyria, Autosomal Recessive. 2012 Sep 27 [Updated 2017 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100826[↩][↩][↩][↩][↩]
- Ramanujam VS, Anderson KE. Porphyria diagnostics—part 1: a brief overview of the porphyrias. Current Protocols in Human Genetics. 2015;86:17.20.1–17.20.26. doi: 10.1002/0471142905.hg1720s86[↩]
- About Porphyria. https://porphyriafoundation.org/for-patients/about-porphyria/[↩][↩]
- Bissell DM, Wang B. Acute hepatic porphyria. Journal of Clinical and Translational Hepatology. 2015;3(1):17–26. doi: 10.14218/JCTH.2014.00039[↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Heme and Bilirubin Metabolism. https://themedicalbiochemistrypage.org/heme-and-bilirubin-metabolism/[↩]
- Marcacci, M., Ricci, A., Cuoghi, C. et al. Challenges in diagnosis and management of acute hepatic porphyrias: from an uncommon pediatric onset to innovative treatments and perspectives. Orphanet J Rare Dis 17, 160 (2022). https://doi.org/10.1186/s13023-022-02314-9[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩][↩][↩][↩]
- Kakoullis L, Louppides S, Papachristodoulou E, Panos G. Porphyrias and photosensitivity: pathophysiology for the clinician. Postgrad Med. 2018 Nov;130(8):673-686. doi: 10.1080/00325481.2018.1533380[↩][↩]
- Leaf RK, Dickey AK. Porphyria cutanea tarda: a unique iron-related disorder. Hematology Am Soc Hematol Educ Program. 2024 Dec 6;2024(1):450-456. doi: 10.1182/hematology.2024000664[↩][↩][↩][↩][↩]
- Usta Atmaca H, Akbas F. Porphyria cutanea tarda: a case report. J Med Case Rep. 2019 Jan 21;13(1):17. doi: 10.1186/s13256-018-1956-9[↩]
- DeMaria BL, Franke AJ. Porphyria Cutanea Tarda in a Patient With Hereditary Hemochromatosis: A Complex Overlap Disorder. Cureus. 2024 Nov 20;16(11):e74091. doi: 10.7759/cureus.74091[↩]
- Baravelli CM, Aarsand AK, Sandberg S, Tollånes MC. Porphyria cutanea tarda and patterns of long-term sick leave and disability pension: a 24-year nationwide matched-cohort study. Orphanet J Rare Dis. 2022 Feb 22;17(1):72. doi: 10.1186/s13023-022-02201-3. Erratum in: Orphanet J Rare Dis. 2022 May 4;17(1):180. doi: 10.1186/s13023-022-02329-2[↩]
- Bonkovsky HL, Rudnick SP, Ma CD, Overbey JR, Wang K, Faust D, Hallberg C, Hedstrom K, Naik H, Moghe A, Anderson KE. Ledipasvir/Sofosbuvir Is Effective as Sole Treatment of Porphyria Cutanea Tarda with Chronic Hepatitis C. Dig Dis Sci. 2023 Jun;68(6):2738-2746. doi: 10.1007/s10620-023-07859-8[↩]
- Awad A, Nirenberg A, Sinclair R. Case Report: Treatment of porphyria cutanea tarda with low dose hydroxychloroquine. F1000Res. 2022 Aug 17;11:945. doi: 10.12688/f1000research.124022.1[↩]
- Norman RA. Past and future: porphyria and porphyrins. Skinmed. 2005 Sep-Oct;4(5):287-92. doi: 10.1111/j.1540-9740.2005.03706.x[↩]
- Badenas C, To-Figueras J, Phillips JD, Warby CA, Muñoz C, Herrero C. Identification and characterization of novel uroporphyrinogen decarboxylase gene mutations in a large series of porphyria cutanea tarda patients and relatives. Clin Genet. 2009 Apr;75(4):346-53. doi: 10.1111/j.1399-0004.2009.01153.x[↩]
- Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP. A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2007 Mar 20;104(12):5079-84. doi: 10.1073/pnas.0700547104[↩][↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw–Hill; 2001. pp. 2991–3062.[↩]
- Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135(3):281–292. doi: 10.1111/j.1365-2141.2006.06289.x[↩][↩]
- Shah A, Bhatt H. Porphyria Cutanea Tarda. [Updated 2023 Apr 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563209[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Farrag MS, Mikula I, Richard E, Saudek V, De Verneuil H, Martásek P. Hepatoerythropoietic Porphyria Caused by a Novel Homoallelic Mutation in Uroporphyrinogen Decarboxylase Gene in Egyptian Patients. Folia Biol (Praha) 2015;61(6):219–226. doi: 10.14712/fb2015061060219[↩]
- Mykletun M, Aarsand AK, Støle E, Villanger JH, Tollånes MC, Baravelli C, Sandberg S. Porphyrias in Norway. Tidsskr Nor Laegeforen. 2014 Apr 29;134(8):831-6. English, Norwegian. doi: 10.4045/tidsskr.13.0649[↩]
- Bulaj ZJ, Phillips JD, Ajioka RS, Franklin MR, Griffen LM, Guinee DJ, Edwards CQ, Kushner JP. Hemochromatosis genes and other factors contributing to the pathogenesis of porphyria cutanea tarda. Blood. 2000 Mar 1;95(5):1565-71. https://doi.org/10.1182/blood.V95.5.1565.005k42_1565_1571[↩][↩]
- Frank J, Poblete-Gutiérrez P. Porphyria cutanea tarda–when skin meets liver. Best Pract Res Clin Gastroenterol. 2010 Oct;24(5):735-45. doi: 10.1016/j.bpg.2010.07.002[↩][↩]
- Rudnick S, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Familial Porphyria Cutanea Tarda. 2013 Jun 6 [Updated 2022 Jun 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK143129[↩][↩][↩][↩][↩][↩][↩]
- Handler NS, Handler MZ, Stephany MP, Handler GA, Schwartz RA. Porphyria cutanea tarda: an intriguing genetic disease and marker. Int J Dermatol. 2017 Jun;56(6):e106-e117. doi: 10.1111/ijd.13580[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012 Jul;32(6):880-93. doi: 10.1111/j.1478-3231.2012.02794.x[↩][↩][↩]
- Ellervik C, Birgens H, Tybjaerg-Hansen A, Nordestgaard BG. Hemochromatosis genotypes and risk of 31 disease endpoints: meta-analyses including 66,000 cases and 226,000 controls. Hepatology. 2007 Oct;46(4):1071-80. doi: 10.1002/hep.21885[↩][↩]
- Ratnaike S, Blake D, Campbell D, Cowen P, Varigos G. Plasma ferritin levels as a guide to the treatment of porphyria cutanea tarda by venesection. Australas J Dermatol. 1988 Apr;29(1):3-8. doi: 10.1111/j.1440-0960.1988.tb01216.x[↩][↩][↩]
- Rocchi E, Gibertini P, Cassanelli M, Pietrangelo A, Borghi A, Ventura E. Serum ferritin in the assessment of liver iron overload and iron removal therapy in porphyria cutanea tarda. J Lab Clin Med. 1986 Jan;107(1):36-42.[↩][↩][↩]
- Balogun O, Nejak-Bowen K. Understanding Hepatic Porphyrias: Symptoms, Treatments, and Unmet Needs. Semin Liver Dis. 2024 May;44(2):209-225. doi: 10.1055/s-0044-1787076[↩][↩][↩][↩][↩][↩]
- Porphyria Cutanea Tarda. https://emedicine.medscape.com/article/1103643-overview#a7[↩]
- Egger NG, Goeger DE, Payne DA, Miskovsky EP, Weinman SA, Anderson KE. Porphyria cutanea tarda: multiplicity of risk factors including HFE mutations, hepatitis C, and inherited uroporphyrinogen decarboxylase deficiency. Dig Dis Sci. 2002 Feb;47(2):419-26. doi: 10.1023/a:1013746828074[↩]
- Cruz-Rojo J, Fontanellas A, Morán-Jiménez MJ, Navarro-Ordóñez S, García-Bravo M, Méndez M, Muñoz-Rivero MC, de Salamanca RE. Precipitating/aggravating factors of porphyria cutanea tarda in Spanish patients. Cell Mol Biol (Noisy-le-grand). 2002 Dec;48(8):845-52.[↩][↩][↩]
- Jalil S, Grady JJ, Lee C, Anderson KE. Associations among behavior-related susceptibility factors in porphyria cutanea tarda. Clin Gastroenterol Hepatol. 2010 Mar;8(3):297-302, 302.e1. doi: 10.1016/j.cgh.2009.11.017[↩]
- HFE gene. https://medlineplus.gov/genetics/gene/hfe/[↩][↩][↩][↩]
- Porter JL, Rawla P. Hemochromatosis. [Updated 2024 Oct 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430862[↩]
- Singal AK. Porphyria cutanea tarda: Recent update. Mol Genet Metab. 2019 Nov;128(3):271-281. doi: 10.1016/j.ymgme.2019.01.004[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bonkovsky HL, Lambrecht RW, Shan Y. Iron as a co-morbid factor in nonhemochromatotic liver disease. Alcohol. 2003 Jun;30(2):137-44. doi: 10.1016/s0741-8329(03)00127-7[↩]
- Bonkovsky HL, Poh-Fitzpatrick M, Pimstone N, Obando J, Di Bisceglie A, Tattrie C, Tortorelli K, LeClair P, Mercurio MG, Lambrecht RW. Porphyria cutanea tarda, hepatitis C, and HFE gene mutations in North America. Hepatology. 1998 Jun;27(6):1661-9. doi: 10.1002/hep.510270627[↩][↩][↩]
- Barton JC, Parker CJ. HFE-Related Hemochromatosis. 2000 Apr 3 [Updated 2024 Apr 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1440[↩][↩][↩][↩][↩]
- Hereditary hemochromatosis. https://medlineplus.gov/genetics/condition/hereditary-hemochromatosis/[↩][↩][↩]
- Muñoz-Santos C, Guilabert A, Moreno N, To-Figueras J, Badenas C, Darwich E, Herrero C. Familial and sporadic porphyria cutanea tarda: clinical and biochemical features and risk factors in 152 patients. Medicine (Baltimore). 2010 Mar;89(2):69-74. doi: 10.1097/MD.0b013e3181d50928[↩][↩][↩][↩]
- McColl KE, Moore MR, Thompson GG, Goldberg A. Abnormal haem biosynthesis in chronic alcoholics. Eur J Clin Invest. 1981 Dec;11(6):461-8. doi: 10.1111/j.1365-2362.1981.tb02014.x[↩]
- Fontanellas A, Martínez-Fresno M, Garrido-Astray MC, Perucho T, Morán-Jiménez MJ, García-Bravo M, Méndez M, Poblete-Gutiérrez P, Frank J, Henriques-Gil N, de Salamanca RE. Smoking but not homozygosity for CYP1A2 g-163A allelic variant leads to earlier disease onset in patients with sporadic porphyria cutanea tarda. Exp Dermatol. 2010 Aug;19(8):e326-8. doi: 10.1111/j.1600-0625.2009.01040.x[↩]
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017 Aug 31;377(9):862-872. doi: 10.1056/NEJMra1608634[↩][↩][↩][↩]
- Quansah R, Cooper CJ, Said S, Bizet J, Paez D, Hernandez GT. Hepatitis C- and HIV-induced porphyria cutanea tarda. Am J Case Rep. 2014 Jan 21;15:35-40. doi: 10.12659/AJCR.889955[↩]
- Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sakai A, Okuda M, Hidaka I, Okita K, Sakaida I. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008 Jan;134(1):226-38. doi: 10.1053/j.gastro.2007.10.011[↩][↩]
- Cruz MJ, Alves S, Baudrier T, Azevedo F. Porphyria cutanea tarda induced by tamoxifen. Dermatol Online J. 2010 Sep 15;16(9):2. https://doi.org/10.5070/D38xm7n81b[↩]
- Bulaj ZJ, Franklin MR, Phillips JD, Miller KL, Bergonia HA, Ajioka RS, Griffen LM, Guinee DJ, Edwards CQ, Kushner JP. Transdermal estrogen replacement therapy in postmenopausal women previously treated for porphyria cutanea tarda. J Lab Clin Med. 2000 Dec;136(6):482-8. doi: 10.1067/mlc.2000.111024[↩][↩][↩][↩]
- Roenigk HH, Gottlob ME. Estrogen-Induced Porphyria Cutanea Tarda: Report of Three Cases. Arch Dermatol. 1970;102(3):260–266. doi:10.1001/archderm.1970.04000090022004[↩]
- Ajioka RS, Phillips JD, Weiss RB, Dunn DM, Smit MW, Proll SC, Katze MG, Kushner JP. Down-regulation of hepcidin in porphyria cutanea tarda. Blood. 2008 Dec 1;112(12):4723-8. doi: 10.1182/blood-2008-02-138222[↩][↩]
- Gibson GE, McEvoy MT. Coexistence of lupus erythematosus and porphyria cutanea tarda in fifteen patients. J Am Acad Dermatol. 1998 Apr;38(4):569-73. doi: 10.1016/s0190-9622(98)70119-7[↩]
- Rodrigues N, Caeiro F, Santana A, Mendes T, Lopes L. Porphyria Cutanea Tarda in a Patient with End-Stage Renal Disease: A Case of Successful Treatment with Deferoxamine and Ferric Carboxymaltose. Case Rep Nephrol. 2017;2017:4591871. doi: 10.1155/2017/4591871[↩]
- Christiansen AL, Bygum A, Hother-Nielsen O, Rasmussen LM. Diagnosing diabetes mellitus in patients with porphyria cutanea tarda. Int J Dermatol. 2018 Jul;57(7):763-769. doi: 10.1111/ijd.13938[↩]
- McKenna DB, Browne M, O’Donnell R, Murphy GM. Porphyria cutanea tarda and hematologic malignancy–a report of 4 cases. Photodermatol Photoimmunol Photomed. 1997 Aug;13(4):143-6. doi: 10.1111/j.1600-0781.1997.tb00218.x[↩]
- Dubart A, Mattei MG, Raich N, Beaupain D, Romeo PH, Mattei JF, Goossens M. Assignment of human uroporphyrinogen decarboxylase (URO-D) to the p34 band of chromosome 1. Hum Genet. 1986 Jul;73(3):277-9. doi: 10.1007/BF00401245[↩]
- Romana M, Dubart A, Beaupain D, Chabret C, Goossens M, Romeo PH. Structure of the gene for human uroporphyrinogen decarboxylase. Nucleic Acids Res. 1987 Sep 25;15(18):7343-56. https://pmc.ncbi.nlm.nih.gov/articles/instance/306252/pdf/nar00262-0144.pdf[↩]
- Felsher BF, Carpio NM, Engleking DW, Nunn AT. Decreased hepatic uroporphyrinogen decarboxylase activity in porphyria cutanea tarda. N Engl J Med. 1982 Apr 1;306(13):766-9. doi: 10.1056/NEJM198204013061302[↩]
- Phillips JD. Heme biosynthesis and the porphyrias. Mol Genet Metab. 2019 Nov;128(3):164-177. doi: 10.1016/j.ymgme.2019.04.008[↩][↩][↩]
- Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP. The uroporphomethene inhibitor causitive for porphyria cutanea tarda (PCT) is generated by oxidation of hydroxymethylbilane (HMB). Blood. 2008; 112(11):3454.[↩]
- Phillips JD, Whitby FG, Kushner JP, Hill CP. Structural basis for tetrapyrrole coordination by uroporphyrinogen decarboxylase. EMBO J. 2003 Dec 1;22(23):6225-33. doi: 10.1093/emboj/cdg606[↩][↩]
- Danton M, Lim CK. Porphomethene inhibitor of uroporphyrinogen decarboxylase: analysis by high-performance liquid chromatography/electrospray ionization tandem mass spectrometry. Biomed Chromatogr. 2007 Jul;21(7):661-3. doi: 10.1002/bmc.860[↩]
- Poh-Fitzpatrick MB. Pathogenesis and treatment of photocutaneous manifestations of the porphyrias. Semin Liver Dis. 1982 May;2(2):164-76. doi: 10.1055/s-2008-1040706[↩][↩]
- Phillips JD, Jackson LK, Bunting M, Franklin MR, Thomas KR, Levy JE, Andrews NC, Kushner JP. A mouse model of familial porphyria cutanea tarda. Proc Natl Acad Sci U S A. 2001 Jan 2;98(1):259-64. doi: 10.1073/pnas.98.1.259[↩]
- Sarkany RP. The management of porphyria cutanea tarda. Clin Exp Dermatol. 2001 May;26(3):225-32. doi: 10.1046/j.1365-2230.2001.00825.x[↩]
- Di Pierro E, De Canio M, Mercadante R, Savino M, Granata F, Tavazzi D, Nicolli AM, Trevisan A, Marchini S, Fustinoni S. Laboratory Diagnosis of Porphyria. Diagnostics (Basel). 2021 Jul 26;11(8):1343. doi: 10.3390/diagnostics11081343[↩][↩]
- Elder GH. Differentiation of porphyria cutanea tarda symptomatica from other types of porphyria by measurement of isocoproporphyrin in faeces. J Clin Pathol. 1975 Aug;28(8):601-7. https://pmc.ncbi.nlm.nih.gov/articles/instance/475786/pdf/jclinpath00142-0001.pdf[↩]
- Wang Y, Gatti P, Sadílek M, Scott CR, Turecek F, Gelb MH. Direct assay of enzymes in heme biosynthesis for the detection of porphyrias by tandem mass spectrometry. Uroporphyrinogen decarboxylase and coproporphyrinogen III oxidase. Anal Chem. 2008 Apr 1;80(7):2599-605. doi: 10.1021/ac702130n[↩]
- Yasuda M, Chen B, Desnick RJ. Recent advances on porphyria genetics: Inheritance, penetrance & molecular heterogeneity, including new modifying/causative genes. Mol Genet Metab. 2019 Nov;128(3):320-331. doi: 10.1016/j.ymgme.2018.11.012[↩]
- Bleasel NR, Varigos GA. Porphyria cutanea tarda. Australas J Dermatol. 2000 Nov;41(4):197-206; quiz 207-8. doi: 10.1046/j.1440-0960.2000.00437.x[↩][↩]
- Easterbrook M. An ophthalmological view on the efficacy and safety of chloroquine versus hydroxychloroquine. J Rheumatol. 1999 Sep;26(9):1866-8.[↩]
- Salameh H, Sarairah H, Rizwan M, Kuo YF, Anderson KE, Singal AK. Relapse of porphyria cutanea tarda after treatment with phlebotomy or 4-aminoquinoline antimalarials: a meta-analysis. Br J Dermatol. 2018 Dec;179(6):1351-1357. doi: 10.1111/bjd.16741[↩]
- Porphyria Cutanea Tarda. https://emedicine.medscape.com/article/1103643-overview#a2[↩]
- Gisbert JP, García-Buey L, Alonso A, Rubio S, Hernández A, Pajares JM, García-Díez A, Moreno-Otero R. Hepatocellular carcinoma risk in patients with porphyria cutanea tarda. Eur J Gastroenterol Hepatol. 2004 Jul;16(7):689-92. doi: 10.1097/01.meg.0000108318.52416.c9[↩]
- Rudnick S, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Hepatoerythropoietic Porphyria. 2013 Oct 31 [Updated 2022 Dec 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK169003[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Kaya Ç, Heydari A, Kızılay O, Kızılay O, Sezgin G. Hepatoerythropoietic Porphyria with Coexisting BTD And CNGB1 Genetic Mutations: A First Case Report. Eur J Case Rep Intern Med. 2025 Jan 25;12(3):005198. doi: 10.12890/2025_005198[↩]
- Cantatore-Francis JL, Cohen-Pfeffer J, Balwani M, Kahn P, Lazarus HM, Desnick RJ, Schaffer JV. Hepatoerythropoietic porphyria misdiagnosed as child abuse: cutaneous, arthritic, and hematologic manifestations in siblings with a novel UROD mutation. Arch Dermatol. 2010 May;146(5):529-33. doi: 10.1001/archdermatol.2010.89[↩][↩][↩][↩][↩][↩][↩][↩]
- Hepatoerythropoietic Porphyria (HEP). https://porphyriafoundation.org/for-patients/types-of-porphyria/hep/[↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CS, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th. New York, NY: McGraw-Hill; 2001. pp. 2961–3062.[↩]
- de Verneuil H, Grandchamp B, Beaumont C, Picat C, Nordmann Y. Uroporphyrinogen decarboxylase structural mutant (Gly281—-Glu) in a case of porphyria. Science. 1986 Nov 7;234(4777):732-4. doi: 10.1126/science.3775362[↩]
- Smith SG. Hepatoerythropoietic porphyria. Semin Dermatol. 1986;5(2):125–137.[↩][↩][↩][↩]
- Ged C, Ozalla D, Herrero C, et al. Description of a New Mutation in Hepatoerythropoietic Porphyria and Prenatal Exclusion of a Homozygous Fetus. Arch Dermatol. 2002;138(7):957–960. doi:10.1001/archderm.138.7.957[↩][↩][↩]
- 92 – Congenital and Hereditary Disorders of the Skin. Avery’s Diseases of the Newborn (Eleventh Edition) 2024, Pages 1332-1346, 1346.e1-1346.e2 https://doi.org/10.1016/B978-0-323-82823-9.00092-1[↩]
- Berenguer J, Blasco J, Cardenal C, Pujol T, Cruces Prado MJ, Herrero C, Mascaró JM, de la Torre C, Mercader JM. Hepatoerythropoietic porphyria: neuroimaging findings. AJNR Am J Neuroradiol. 1997 Sep;18(8):1557-60. https://pmc.ncbi.nlm.nih.gov/articles/instance/8338136/pdf/9296199.pdf[↩][↩][↩]
- Kaya Ç, Heydari A, Kızılay O, Kızılay O, Sezgin G. Hepatoerythropoietic Porphyria with Coexisting BTD And CNGB1 Genetic Mutations: A First Case Report. Eur J Case Rep Intern Med. 2025 Jan 25;12(3):005198. doi: 10.12890/2025_005198 [↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias, in: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G (Eds.), The Online Metabolic and Molecular Bases of Inherited Disease, The McGraw-Hill Companies, Inc., New York, NY, 2014.[↩]
- Pin˜ol J, Herrero C, Almeida J, et al. Porphyrie he´ pato-erythro-cytaire: une nouvelle forme de porphyrie. Ann Dermatol Venereol 1975;102:129 –136[↩]
- Elder GH. Hepatic porphyrias in children. J Inherit Metab Dis. 1997 Jun;20(2):237-46. doi: 10.1023/a:1005313024076[↩][↩][↩][↩][↩][↩][↩]
- Biesecker LG, Adam MP, Alkuraya FS, et al. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet. 2021 Jan 7;108(1):8-15. doi: 10.1016/j.ajhg.2020.11.013[↩]
- de Verneuil H, Beaumont C, Deybach JC, Nordmann Y, Sfar Z, Kastally R. Enzymatic and immunological studies of uroporphyrinogen decarboxylase in familial porphyria cutanea tarda and hepatoerythropoietic porphyria. Am J Hum Genet. 1984 May;36(3):613-22. https://pmc.ncbi.nlm.nih.gov/articles/instance/1684444/pdf/ajhg00165-0121.pdf[↩][↩][↩]
- Phillips JD, Whitby FG, Stadtmueller BM, Edwards CQ, Hill CP, Kushner JP. Two novel uroporphyrinogen decarboxylase (URO-D) mutations causing hepatoerythropoietic porphyria (HEP) Transl Res. 2007;149(2):85–91. doi: 10.1016/j.trsl.2006.08.006[↩]
- Granata BX, Parera VE, Melito VA, Teijo MJ, Batlle AM, Rossetti MV. The very first description of a patient with hepatoerythropoietic porphyria in Argentina. Biochemical and molecular studies. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):61-5.[↩][↩]
- Hepatoerythropoietic Porphyria. https://rarediseases.org/rare-diseases/hepatoerythropoietic-porphyria/[↩][↩][↩][↩][↩]
- Weiss Y, Chen B, Yasuda M, Nazarenko I, Anderson KE, Desnick RJ. Porphyria cutanea tarda and hepatoerythropoietic porphyria: Identification of 19 novel uroporphyrinogen III decarboxylase mutations. Mol Genet Metab. 2019 Nov;128(3):363-366. doi: 10.1016/j.ymgme.2018.11.013[↩]
- Aarsand AK, Boman H, Sandberg S. Familial and sporadic porphyria cutanea tarda: characterization and diagnostic strategies. Clin Chem. 2009 Apr;55(4):795-803. doi: 10.1373/clinchem.2008.117432[↩]
- Triviboonvanich S, Junnu S, Srisawat C, Silpa-archa N. Late-onset hepatoerythropoietic porphyria presenting with facial deformities and erythrodontia. Thai J Dermatol. 2019;35:73–80.[↩]
- Harrison-Findik DD, Klein E, Crist C, Evans J, Timchenko N, Gollan J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology. 2007 Dec;46(6):1979-85. doi: 10.1002/hep.21895[↩]
- Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, Fein E, Andriopoulos B, Pantopoulos K, Gollan J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006 Aug 11;281(32):22974-82. doi: 10.1074/jbc.M602098200[↩]
- Fujita N, Sugimoto R, Motonishi S, Tomosugi N, Tanaka H, Takeo M, Iwasa M, Kobayashi Y, Hayashi H, Kaito M, Takei Y. Patients with chronic hepatitis C achieving a sustained virological response to peginterferon and ribavirin therapy recover from impaired hepcidin secretion. J Hepatol. 2008 Nov;49(5):702-10. doi: 10.1016/j.jhep.2008.05.014[↩]
- Beer K, Applebaum D, Nousari C. Pseudoporphyria: discussion of etiologic agents. J Drugs Dermatol. 2014 Aug;13(8):990-2. https://jddonline.com/articles/pseudoporphyria-discussion-of-etiologic-agents-S1545961614P0990X/[↩]
- Piñol Aguadé J, Herrero C, Almeida J, Castells Mas A, Ferrando J, De Asprer J, Palou A, Giménez A. Porphyrie hépato-érythrocytaire. Une nouvelle forme de porphyrie [Hepato-erythrocytic porphyria. A new type of porphyria]. Ann Dermatol Syphiligr (Paris). 1975;102(2):129-36. French.[↩]
- Herrick AL, McColl KE, Moore MR, Cook A, Goldberg A. Controlled trial of haem arginate in acute hepatic porphyria. Lancet. 1989;1:1295–1297. doi: 10.1016/s0140-6736(89)92688-3[↩]
- Petersen CS, Thomsen K. High-dose hydroxychloroquine treatment of porphyria cutanea tarda. J Am Acad Dermatol. 1992;26:614–619. doi: 10.1016/0190-9622(92)70090-3[↩]
- Gunther WW. The porphyrias and erythropoietic protoporphyria: an unusual case. Australas J Dermatol. 1967;9(1):23–30. doi: 10.1111/j.1440-0960.1967.tb01147.x[↩]
- Ged C, Ozalla D, Herrero C, et al. Description of a new mutation in hepatoerythropoietic porphyria and prenatal exclusion of a homozygous fetus. Arch Dermatol. 2002;138(7):957–960. doi: 10.1001/archderm.138.7.957[↩]
- Remenyik É, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol. 2008;33(5):602–605. doi: 10.1111/j.1365-2230.2008.02734.x[↩][↩]
- Czarnecki DB. Hepatoerythropoietic Porphyria. Arch Dermatol. 1980;116(3):307–311. doi:10.1001/archderm.1980.01640270067017[↩]
- Horina JH, Wolf P. Epoetin for severe anemia in hepatoerythropoietic porphyria. N Engl J Med. 2000;342(17):1294–1295. doi: 10.1056/NEJM200004273421717[↩][↩]
- Armstrong DK, Sharpe PC, Chambers CR, Whatley SD, Roberts AG, Elder GH. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol. 2004;151(4):920–923. doi: 10.1111/j.1365-2133.2004.06101.x[↩][↩]
- Moran-Jimenez MJ, Ged C, Romana M, Enriquez De Salamanca R, Taïeb A, Topi G, D’Alessandro L, de Verneuil H. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet. 1996 Apr;58(4):712-21. https://pmc.ncbi.nlm.nih.gov/articles/instance/1914669/pdf/ajhg00017-0069.pdf[↩]
- Palmer RA, Elder GH, Barrett DF, Keohane SG. Homozygous variegate porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene. Br J Dermatol. 2001;144(4):866–869. doi: 10.1046/j.1365-2133.2001.04147.x[↩]
- Elder GH. Hepatic porphyrias in children. J Inher Metab Dis. 1997;20(2):237–246. doi: 10.1023/a:1005313024076[↩]
- Murphy GM. The cutaneous porphyrias: a review. The British Photodermatology Group. Br J Dermatol. 1999;140(4):573–581. doi: 10.1046/j.1365-2133.1999.02754.x[↩]
- Parsons JL, Sahn EE, Holden KR, Pai GS. Neurologic disease in a child with hepatoerythropoietic porphyria. Pediatr Dermatol. 1994;11(3):216–221. doi: 10.1111/j.1525-1470.1994.tb00589.x[↩][↩]
- Dickey AK, Naik H, Keel SB, et al. Porphyrias Consortium of the Rare Diseases Clinical Research Network. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol. 2023 Dec;89(6):1227-1237. doi: 10.1016/j.jaad.2022.08.036[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Minder AE, Kluijver LG, Barman-Aksözen J, Minder EI, Langendonk JG. Erythropoietic protoporphyrias: Pathogenesis, diagnosis and management. Liver Int. 2025 Jan;45(1):e16027. doi: 10.1111/liv.16027. Epub 2024 Jul 16. Erratum in: Liver Int. 2024 Dec;44(12):3292. doi: 10.1111/liv.16104[↩]
- Leaf RK, Dickey AK. How I treat erythropoietic protoporphyria and X-linked protoporphyria. Blood. 2023 Jun 15;141(24):2921-2931. doi: 10.1182/blood.2022018688[↩][↩][↩]
- Balwani M, Naik H, Anderson KE, et al. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol 2017;153(8):789–796. doi: 10.1001/jamadermatol.2017.1557[↩]
- Kluijver LG, Wensink D, Wagenmakers MAEM, Huidekoper HH, Witters P, Rymen D, Langendonk JG. Quality of life in children with erythropoietic protoporphyria: a case-control study. J Dermatol. 2024 Aug;51(8):1068-1078. doi: 10.1111/1346-8138.17348[↩]
- Lecha M, Puy H, Deybach JC. Erythropoietic protoporphyria. Orphanet J Rare Dis. 2009 Sep 10;4:19. doi: 10.1186/1750-1172-4-19[↩][↩][↩][↩][↩][↩]
- Ardalan ZS, Chandran S, Vasudevan A, Angus PW, Grigg A, He S, Macdonald GA, Strasser SI, Tate CJ, Kennedy GA, Testro AG, Gow PJ. Management of Patients With Erythropoietic Protoporphyria-Related Progressive Liver Disease. Liver Transpl. 2019 Nov;25(11):1620-1633. doi: 10.1002/lt.25632[↩]
- Ahmed jan N, Masood S. Erythropoietic Protoporphyria. [Updated 2023 Feb 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK563141[↩][↩][↩][↩][↩][↩][↩]
- Erythropoietic Protoporphyria and X-Linked Protoporphyria. https://rarediseases.org/rare-diseases/erythropoietic-protoporphyria[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- McNeil MM, Nahhas AF, Braunberger TL, Hamzavi IH. Afamelanotide in the Treatment of Dermatologic Disease. Skin Therapy Lett. 2018 Nov;23(6):6-10. https://www.skintherapyletter.com/dermatology/afamelanotide[↩][↩][↩]
- Magnus IA, Jarret A, Prankerd TAJ, Rimington C. Erythropoietic protoporphyria: a new porphyria syndrome with solar urticaria due to protoporphyrinæmia. Lancet. 1961;278:574–581. doi: 10.1016/s0140-6736(61)92427-8[↩]
- Cox TM. Protoporpnhyria. In: Kadish KM, Smith KM, Guilard R, editor. The Porphyrin Handbook Medical Aspects of Poprhyrias, Chap 90. Vol. 14. Academic Press. San Diego; 2003. pp. 121–149.[↩]
- Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–14. doi: 10.1016/j.ajhg.2008.08.003[↩]
- Levy C, Naik H, Overbey J, et al. Porphyrias Consortium of the Rare Diseases Clinical Research Network. Liver involvement in a large cohort of patients with erythropoietic protoporphyria or X-linked protoporphyria. Hepatol Commun. 2025 Feb 19;9(3):e0657. doi: 10.1097/HC9.0000000000000657[↩]
- Levy C, Dickey AK, Wang B, Thapar M, et al. Porphyrias Consortium of the Rare Diseases Clinical Network. Evidence-based consensus guidelines for the diagnosis and management of protoporphyria-related liver dysfunction in erythropoietic protoporphyria and X-linked protoporphyria. Hepatology. 2024 Mar 1;79(3):731-743. doi: 10.1097/HEP.0000000000000546[↩]
- FECH gene. https://medlineplus.gov/genetics/gene/fech[↩][↩]
- Snast I, Kaftory R, Sherman S, Edel Y, Hodak E, Levi A, Lapidoth M. Acquired erythropoietic protoporphyria: A systematic review of the literature. Photodermatol Photoimmunol Photomed. 2020 Jan;36(1):29-33. doi: 10.1111/phpp.12501[↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet 2008;83(3):408–414. doi: 10.1016/j.ajhg.2008.08.003[↩][↩][↩][↩]
- ALAS2 gene. https://medlineplus.gov/genetics/gene/alas2[↩][↩][↩][↩][↩][↩]
- Balwani M. Erythropoietic Protoporphyria and X-Linked Protoporphyria: pathophysiology, genetics, clinical manifestations, and management. Mol Genet Metab. 2019 Nov;128(3):298-303. doi: 10.1016/j.ymgme.2019.01.020[↩][↩][↩][↩][↩][↩][↩]
- Madu AE, Whittaker SJ. Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol 2006;26(7):687–688. doi: 10.1080/01443610600930670[↩]
- Jacquemyn Y Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol 2003;23(2):196. doi: 10.1080/0144361031000074817[↩]
- Nevins EG, Wijesiriwardana A. Erythropoietic protoporphyria in pregnancy. J Obstet Gynaecol Published online 2020:1–2. doi: 10.1080/01443615.2020.1777954[↩]
- Bewley AP, Keefe M, White JE. Erythropoietic protoporphyria improving during pregnancy. Br J Dermatol 1998;139(1):145–147. doi: 10.1046/j.1365-2133.1998.02333.x[↩]
- Poh-Fitzpatrick MB. Human protoporphyria: reduced cutaneous photosensitivity and lower erythrocyte porphyrin levels during pregnancy. J Am Acad Dermatol 1997;36(1):40–43. doi: 10.1016/s0190-9622(97)70323-2[↩]
- Heerfordt IM, Wulf HC. Patients with erythropoietic protoporphyria have reduced erythrocyte protoporphyrin IX from early in pregnancy. Br J Dermatol 2017;177(3):e38–e40. doi: 10.1111/bjd.15228[↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ: Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Basis of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2991-3062.[↩][↩]
- Wahlin S, Floderus Y, Stål P, Harper P. Erythropoietic protoporphyria in Sweden: demographic, clinical, biochemical and genetic characteristics. J Intern Med. 2011 Mar;269(3):278-88. doi: 10.1111/j.1365-2796.2010.02236.x[↩][↩][↩]
- Went L, Klasen EC. Genetic aspects of erythropoietic protoporphyria. Ann Hum Genet. 1984;48:105–117. doi: 10.1111/j.1469-1809.1984.tb01006.x[↩]
- Elder GH, Smith SG, Smyth SJ. Laboratory investigation of the porphyrias. Ann Clin Biochem. 1990;27:395–412. doi: 10.1177/000456329002700501[↩][↩]
- Balwani M, Bloomer R, Desnick R, Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network . In: GeneReviews. Adam MP, Mirzaa GM, Pagon RA, editors. University of Seattle; 2017. Erythropoietic protoporphyria, autosomal recessive; pp. 1993–2023.[↩]
- Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The incidence of inherited porphyrias in Europe. J Inherit Metab Dis. 2013;36(5):849–857. doi: 10.1007/s10545-012-9544-4[↩]
- Dickey AK, Quick C, Ducamp S, et al. Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genet Med. 2021;23(1):140–148. doi: 10.1038/s41436-020-00951-8[↩]
- Holme SA, Anstey AV, Finlay AY, Elder GH, Badminton MN. Erythropoietic protoporphyria in the U.K.: clinical features and effect on quality of life. Br J Dermatol. 2006;155:574–581. doi: 10.1111/j.1365-2133.2006.07472.x[↩][↩]
- Wulf HC, Nissen CV, Philipsen PA. Inactivation of protoporphyrin IX in erythrocytes in patients with erythropoietic protoporphyria: A new treatment modality. Photodiagnosis Photodyn Ther. 2020 Mar;29:101582. doi: 10.1016/j.pdpdt.2019.101582[↩]
- Gou EW, Balwani M, Bissell DM, et al. Pitfalls in erythrocyte protoporphyrin measurement for diagnosis and monitoring of protoporphyrias. Clin Chem. 2015;61(12):1453–1456. doi: 10.1373/clinchem.2015.245456[↩][↩][↩][↩][↩]
- Minder EI, Schneider‐Yin X. Laboratory Guide to the Methods in Biochemical Genetics. Springer Berlin Heidelberg; 2008.[↩]
- Ericson MB, Grapengiesser S, Gudmundson F, Wennberg AM, Larkö O, Moan J, Rosén A. A spectroscopic study of the photobleaching of protoporphyrin IX in solution. Lasers Med Sci. 2003;18(1):56-62. doi: 10.1007/s10103-002-0254-2[↩][↩]
- Holme SA, Anstey A v, Badminton MN, Elder GH. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br J Dermatol 2008;159(1):211–213. doi: 10.1111/j.1365-2133.2008.08616.x[↩][↩]
- Spelt JM, de Rooij FW, Wilson JH, Zandbergen AA. Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis 2010;33:S1–4. doi: 10.1007/s10545-008-1037-0[↩][↩]
- Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl 2008;14(9):1340–1346. doi: 10.1002/lt.21527[↩][↩]
- Levoska MA, Griffith JL, Nagai S, Collins K, Lim HW. A multi-disciplinary approach utilizing filters for surgical procedures in erythropoietic protoporphyria. J Am Acad Dermatol 2020;83:e329–330. doi: 10.1016/j.jaad.2020.02.024[↩][↩]
- Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cellular and Molecular Biology 2009;55(1):84–97. doi: 10.1170/T841[↩]
- Mathews-Roth MM, Kass EH, Fitzpatrick TB, Pathak MA, Harber LC. Phototesting as an objective measure of improvement in erythropoietic protoporphyria. Arch Dermatol 1979;115(12):1391–1392. doi: 10.1001/archderm.1979.04010120001002[↩]
- Mathews-Roth MM, Pathak MA, Fitzpatrick TB, Harber LH, Kass EH. Beta Carotene Therapy for Erythropoietic Protoporphyria and Other Photosensitivity Diseases. Arch Dermatol. 1977;113(9):1229–1232. doi:10.1001/archderm.1977.01640090077011[↩]
- Balwani M, Bonkovsky HL, Belongie KJ, et al. Erythropoietic Protoporphyria: Phase 2 Clinical Trial Results Evaluating the Safety and Effectiveness of Dersimelagon (MT-7117), an Oral MC1R Agonist. Blood 2020;136:51–51. doi: 10.1182/BLOOD-2020-142467[↩]
- Bruguera M, Herrero C. [Liver disease in erythropoietic protoporphyria] Gastroenterol Hepatol. 2005;28:632–636. doi: 10.1016/s0210-5705(05)71529-6[↩][↩]
- Meerman L. Erythropoietic protoporphyria. An overview with emphasis on the liver. Scand J Gastroenterol Suppl. 2000;(232):79-85.[↩]
- Thunell S, Harper P, Brun A. Porphyrins, porphyrin metabolism and porphyrias. IV. Pathophysiology of erythyropoietic protoporphyria–diagnosis, care and monitoring of the patient. Scand J Clin Lab Invest. 2000;60:581–604. doi: 10.1080/003655100448310[↩]
- Todd DJ. Erythropoietic protoporphyria. Br J Dermtol. 1994;131:751–756. doi: 10.1111/j.1365-2133.1994.tb08577.x[↩][↩]
- Bonkovsky HL, Schned AR. Fatal liver failure in protoporphyria. Synergism between ethanol excess and the genetic defect. Gastroenterology. 1986 Jan;90(1):191-201. https://www.gastrojournal.org/article/0016-5085(86)90093-4/pdf?referrer=https%3A%2F%2Fpubmed.ncbi.nlm.nih.gov%2F[↩]
- Anstey AV, Hift RJ. Liver disease in erythropoietic protoporphyria: insights and implications for management. Postgrad Med J. 2007 Dec;83(986):739-48. doi: 10.1136/gut.2006.097576[↩][↩][↩]
- Anstey A v., Hift RJ. Liver disease in erythropoietic protoporphyria: Insights and implications for management. Gut 2007;56(7):1009–1018. doi: 10.1136/gut.2006.097576[↩][↩][↩][↩]
- Whatley SD, Mason NG, Khan M, et al. Autosomal recessive erythropoietic protoporphyria in the United Kingdom: prevalence and relationship to liver disease. J Med Genet 2004;41(8):e105. doi: 10.1136/jmg.2003.016121[↩]
- Coffey A, Leung DH, Quintanilla NM. Erythropoietic protoporphyria: Initial diagnosis with cholestatic liver disease. Pediatrics 2018;141:S445–S450. doi: 10.1542/peds.2016-1625[↩]
- Cripps DJ, Goldfarb SS. Erythropoietic protoporphyria: hepatic cirrhosis. Br J Dermatol 1978;98(3):349–354. doi: 10.1111/j.1365-2133.1978.tb06163.x[↩]
- Khalili MJ, Farahmand F, Hirbod-Mobarakeh A, Yousefi A, Sotoudeh S, Monajemzadeh M, Razaghian A, Rezaei N. Erythropoietic protoporphyria and early onset of cholestasis. Turk J Pediatr. 2012 Nov-Dec;54(6):645-50.[↩]
- Balwani M, Doheny D, Bishop DF, et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med 2013;19(1):26–35. doi: 10.2119/molmed.2012.00340[↩][↩]
- Dickey AK, Quick C, Ducamp S, et al. Evidence in the UK Biobank for the underdiagnosis of erythropoietic protoporphyria. Genetics in Medicine 2021;23(1):140–148. doi: 10.1038/s41436-020-00951-8[↩]
- Gouya L, Martin-Schmitt C, Robreau AM, et al. Contribution of a common single-nucleotide polymorphism to the genetic predisposition for erythropoietic protoporphyria. American Journal of Human Genetics 2006;78(1):2–14. doi: 10.1086/498620[↩]
- Whatley SD, Mason NG, Holme SA, Anstey AV, Elder GH, Badminton MN. Molecular epidemiology of erythropoietic protoporphyria in the U.K. Br J Dermatol. 2010 Mar;162(3):642-6. doi: 10.1111/j.1365-2133.2010.09631.x[↩][↩]
- Balwani M, Naik H, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Overbey JR, Wang B, Singal AK, Liu LU, Desnick RJ. Clinical, Biochemical, and Genetic Characterization of North American Patients With Erythropoietic Protoporphyria and X-linked Protoporphyria. JAMA Dermatol. 2017 Aug 1;153(8):789-796. doi: 10.1001/jamadermatol.2017.1557[↩][↩][↩][↩][↩][↩][↩]
- Yien YY, Ducamp S, van der Vorm LN, Kardon JR, Manceau H, Kannengiesser C, Bergonia HA, Kafina MD, Karim Z, Gouya L, Baker TA, Puy H, Phillips JD, Nicolas G, Paw BH. Mutation in human CLPX elevates levels of δ-aminolevulinate synthase and protoporphyrin IX to promote erythropoietic protoporphyria. Proc Natl Acad Sci U S A. 2017 Sep 19;114(38):E8045-E8052. doi: 10.1073/pnas.1700632114[↩]
- Blagojevic D, Schenk T, Haas O, Zierhofer B, Konnaris C, Trautinger F. Acquired erythropoietic protoporphyria. Ann Hematol. 2010 Jul;89(7):743-4. doi: 10.1007/s00277-009-0859-7[↩]
- Aplin C, Whatley SD, Thompson P, Hoy T, Fisher P, Singer C, Lovell CR, Elder GH. Late-onset erythropoietic porphyria caused by a chromosome 18q deletion in erythroid cells. J Invest Dermatol. 2001 Dec;117(6):1647-9. doi: 10.1046/j.0022-202x.2001.01560.x[↩]
- Suzuki H, Kikuchi K, Fukuhara N, Nakano H, Aiba S. Case of late-onset erythropoietic protoporphyria with myelodysplastic syndrome who has homozygous IVS3-48C polymorphism in the ferrochelatase gene. J Dermatol. 2017 Jun;44(6):651-655. doi: 10.1111/1346-8138.13709[↩]
- Livideanu CB, Ducamp S, Lamant L, Gouya L, Rauzy OB, Deybach JC, Paul C, Puy H, Marguery MC. Late-onset X-linked dominant protoporphyria: an etiology of photosensitivity in the elderly. J Invest Dermatol. 2013 Jun;133(6):1688-90. doi: 10.1038/jid.2012.467[↩]
- Yao YJ, Li SFY. Determination of erythrocyte porphyrins by epi-illumination fluorescence microscope with capillary electrophoresis. J Liq Chromatogr Relat Technol 1996;19(1):1–15. doi: 10.1080/10826079608006285[↩]
- Hennig G, Gruber C, Vogeser M, et al. Dual-wavelength excitation for fluorescence-based quantification of zinc protoporphyrin IX and protoporphyrin IX in whole blood. J Biophotonics 2014;7(7):514–524. doi: 10.1002/jbio.201200228[↩]
- Gou EW, Balwani M, Bissell DM, et al. Pitfalls in Erythrocyte Protoporphyrin Measurement for Diagnosis and Monitoring of Protoporphyrias. Clin Chem 2015;61(12):1453–1456. doi: 10.1373/clinchem.2015.245456[↩]
- Balwani M, Naik H, Anderson KE, et al. Clinical, biochemical, and genetic characterization of North American patients with erythropoietic protoporphyria and X-linked protoporphyria. JAMA Dermatol. 2017;153(8):789–796. doi: 10.1001/jamadermatol.2017.1557[↩][↩]
- Dickey AK, Naik H, Keel SB, et al. Evidence-based consensus guidelines for the diagnosis and management of erythropoietic protoporphyria and X-linked protoporphyria. J Am Acad Dermatol. Published online 27 August 2022 doi: 10.1016/j.jaad.2022.08.036[↩][↩]
- Coffey A, Leung DH, Quintanilla NM. Erythropoietic Protoporphyria: Initial Diagnosis With Cholestatic Liver Disease. Pediatrics. 2018 Apr;141(Suppl 5):S445-S450. doi: 10.1542/peds.2016-1625[↩]
- Drug-induced photosensitivity. https://dermnetnz.org/topics/drug-induced-photosensitivity[↩]
- Hydroa vacciniforme. https://dermnetnz.org/topics/hydroa-vacciniforme[↩]
- Solar urticaria. https://dermnetnz.org/topics/solar-urticaria[↩][↩]
- Polymorphic light eruption. https://dermnetnz.org/topics/polymorphic-light-eruption[↩]
- de Castro Maqueda G, Gutiérrez-Manzanedo JV, González-Montesinos JL, Vaz Pardal C, Rivas Ruiz F, de Troya Martín M. Sun Exposure and Photoprotection: Habits, Knowledge and Attitudes Among Elite Kitesurfers. J Cancer Educ. 2022 Jun;37(3):517-523. doi: 10.1007/s13187-020-01838-7[↩]
- Edel Y, Mamet R, Snast I, Kaftory R, Mazor S, Hodak E, Lapidoth M, Elis A, Molad Y, Levi A. Epidemiology of cutaneous porphyria in Israel: a nationwide cohort study. J Eur Acad Dermatol Venereol. 2020 Jan;34(1):184-187. doi: 10.1111/jdv.15769[↩]
- Solano F. Photoprotection and Skin Pigmentation: Melanin-Related Molecules and Some Other New Agents Obtained from Natural Sources. Molecules 2020;25(7). doi: 10.3390/MOLECULES25071537[↩]
- Langendonk JG, Balwani M, Anderson KE, et al. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015 Jul 2;373(1):48-59. doi: 10.1056/NEJMoa1411481[↩]
- Holme SA, Thomas CL, Whatley SD, Bentley DP, Anstey AV, Badminton MN. Symptomatic response of erythropoietic protoporphyria to iron supplementation. J Am Acad Dermatol. 2007 Jun;56(6):1070-2. doi: 10.1016/j.jaad.2006.11.030[↩][↩]
- Lyoumi S, Abitbol M, Andrieu V, Henin D, Robert E, Schmitt C, Gouya L, de Verneuil H, Deybach JC, Montagutelli X, Beaumont C, Puy H. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood. 2007 Jan 15;109(2):811-8. doi: 10.1182/blood-2006-04-014142[↩][↩]
- Schmidt H, Snitker G, Thomsen K, Lintrup J. Erythropoietic protoporphyria. A clinical study based on 29 cases in 14 families. Arch Dermatol 1974;110(1):58–64. doi: 10.1001/archderm.110.1.58[↩]
- Holme SA, Worwood M, Anstey A v, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood 2007;110(12):4108–4110. doi: 10.1182/blood-2007-04-088120[↩]
- Barman-Aksözen J, Minder EI, Schubiger C, Biolcati G, Schneider-Yin X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol Dis 2015;54(1):71–77. doi: 10.1016/j.bcmd.2014.07.017[↩]
- Lyoumi S, Abitbol M, Andrieu V, et al. Increased plasma transferrin, altered body iron distribution, and microcytic hypochromic anemia in ferrochelatase-deficient mice. Blood 2007;109(2):811–818. doi: 10.1182/blood-2006-04-014142[↩]
- Barman-Aksoezen J, Girelli D, Aurizi C, et al. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J Inherit Metab Dis 2017;40(3):433–441. doi: 10.1007/s10545-017-0017-7[↩]
- Bossi K, Lee J, Schmeltzer P, et al. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest 2015;45(10):1032–1041. doi: 10.1111/eci.12503[↩]
- Bentley DP, Meek EM. Clinical and biochemical improvement following low-dose intravenous iron therapy in a patient with erythropoietic protoporphyria. British Journal of Haematology 2013;163(2):289–291. doi: 10.1111/bjh.12485[↩]
- Delaby C, Lyoumi S, Ducamp S, Martin-Schmitt C, Gouya L, Deybach JC, Beaumont C, Puy H. Excessive erythrocyte PPIX influences the hematologic status and iron metabolism in patients with dominant erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):45-52.[↩]
- Minder EI, Schneider-Yin X, Steurer J, Bachmann LM. A systematic review of treatment options for dermal photosensitivity in erythropoietic protoporphyria. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):84-97.[↩][↩][↩]
- Do KD, Banner BF, Katz E, Szymanski IO, Bonkovsky HL. Benefits of chronic plasmapheresis and intravenous heme-albumin in erythropoietic protoporphyria after orthotopic liver transplantation. Transplantation. 2002 Feb 15;73(3):469-72. doi: 10.1097/00007890-200202150-00024[↩][↩]
- McCullough AJ, Barron D, Mullen KD, Petrelli M, Park MC, Mukhtar H, Bickers DR. Fecal protoporphyrin excretion in erythropoietic protoporphyria: effect of cholestyramine and bile acid feeding. Gastroenterology. 1988 Jan;94(1):177-81. doi: 10.1016/0016-5085(88)90627-0[↩][↩]
- McGuire BM, Bonkovsky HL, Carithers RL Jr, Chung RT, Goldstein LI, Lake JR, Lok AS, Potter CJ, Rand E, Voigt MD, Davis PR, Bloomer JR. Liver transplantation for erythropoietic protoporphyria liver disease. Liver Transpl. 2005 Dec;11(12):1590-6. doi: 10.1002/lt.20620[↩][↩][↩][↩]
- Wahlin S, Stal P, Adam R, Karam V, Porte R, Seehofer D, Gunson BK, Hillingsø J, Klempnauer JL, Schmidt J, Alexander G, O’Grady J, Clavien PA, Salizzoni M, Paul A, Rolles K, Ericzon BG, Harper P; European Liver and Intestine Transplant Association. Liver transplantation for erythropoietic protoporphyria in Europe. Liver Transpl. 2011 Sep;17(9):1021-6. doi: 10.1002/lt.22341[↩][↩]
- Weiss Y, Balwani M, Chen B, Yasuda M, Nazarenko I, Desnick RJ. Congenital erythropoietic porphyria and erythropoietic protoporphyria: Identification of 7 uroporphyrinogen III synthase and 20 ferrochelatase novel mutations. Mol Genet Metab. 2019 Nov;128(3):358-362. doi: 10.1016/j.ymgme.2018.08.015[↩]
- Rand EB, Bunin N, Cochran W, Ruchelli E, Olthoff KM, Bloomer JR. Sequential liver and bone marrow transplantation for treatment of erythropoietic protoporphyria. Pediatrics. 2006 Dec;118(6):e1896-9. doi: 10.1542/peds.2006-0833[↩][↩]
- Wang YM, Gloude NJ, Davies SM, Lucky AW, Nelson AS. Hematopoietic stem cell transplant for erythropoietic porphyrias in pediatric patients. Pediatr Blood Cancer. 2021 Sep;68(9):e29231. doi: 10.1002/pbc.29231[↩]
- Hashmi SK, Harstead E, Sachdev M, Black DD, Clark I, Ortanca I, Triplett BM, Talleur AC. Hematopoietic cell transplant for reversal of liver fibrosis in a pediatric patient with erythropoietic protoporphyria. Pediatr Transplant. 2021 Sep;25(6):e13966. doi: 10.1111/petr.13966[↩][↩]
- Endo Y, Hibi T, Shinoda M, Obara H, Kitago M, Yagi H, Abe Y, Hasegawa Y, Matsubara K, Hori S, Tanaka M, Makiuchi S, Nakano Y, Itano O, Kuroda T, Kitagawa Y. Reappraisal of liver transplantation for erythropoietic protoporphyria: A deadly combination of disease recurrence and biliary complication. Pediatr Transplant. 2022 Jun;26(4):e14261. doi: 10.1111/petr.14261[↩]
- Ricci A, Di Betto G, Bergamini E, Buzzetti E, Corradini E, Ventura P. Iron Metabolism in the Disorders of Heme Biosynthesis. Metabolites. 2022 Aug 31;12(9):819. doi: 10.3390/metabo12090819[↩]
- Rutter KJ, Ashraf I, Cordingley L, Rhodes LE. Quality of life and psychological impact in the photodermatoses: a systematic review. Br J Dermatol. 2020 May;182(5):1092-1102. doi: 10.1111/bjd.18326[↩]
- Balwani M, Desnick R; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. X-Linked Protoporphyria. 2013 Feb 14 [Updated 2019 Nov 27]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK121284[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Levy C, Naik H, Overbey J, Hedstrom K, et al.; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Liver involvement in a large cohort of patients with erythropoietic protoporphyria or X-linked protoporphyria. Hepatol Commun. 2025 Feb 19;9(3):e0657. doi: 10.1097/HC9.0000000000000657[↩]
- Balwani M, Doheny D, Bishop DF, et al. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013;19(1):26–35. doi: 10.2119/molmed.2012.00340[↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83(3):408–414. doi: 10.1016/j.ajhg.2008.08.003[↩]
- Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, Corrigall AV, Meissner PN, Hift RJ, Marsden JT, Ma Y, Mieli-Vergani G, Deybach JC, Puy H. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008 Sep;83(3):408-14. doi: 10.1016/j.ajhg.2008.08.003[↩][↩][↩][↩][↩][↩]
- Manceau H, Gouya L, Puy H. Acute hepatic and erythropoietic porphyrias: from ALA synthases 1 and 2 to new molecular bases and treatments. Curr Opin Hematol. 2017 May;24(3):198-207. doi: 10.1097/MOH.0000000000000330[↩][↩][↩][↩]
- Anderson KE, Sassa S, Bishop DF, and Desnick RJ (2001). Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias In The Metabolic and Molecurlar Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, and Valle D, eds. (New York, McGraw-Hill: ), pp 2961–3062.[↩][↩][↩][↩][↩]
- Whatley SD, Ducamp S, Gouya L, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008 Sep;83(3):408-14. doi: 10.1016/j.ajhg.2008.08.003[↩][↩]
- Poh-Fitzpatrick MB. Erythropoietic protoporphyria. Int J Dermatol. 1978 Jun;17(5):359-69. doi: 10.1111/ijd.1978.17.5.359[↩]
- Poh-Fitzpatrick MB. Porphyrias: photosensitivity and phototherapy. Methods Enzymol. 2000;319:485-93. doi: 10.1016/s0076-6879(00)19045-7[↩][↩]
- Bloomer JR. The liver in protoporphyria. Hepatology. 1988 Mar-Apr;8(2):402-7. doi: 10.1002/hep.1840080235[↩]
- Bloomer JR. Hepatic protoporphyrin metabolism in patients with advanced protoporphyric liver disease. Yale J Biol Med. 1997 Jul-Aug;70(4):323-30. https://pmc.ncbi.nlm.nih.gov/articles/instance/2589331/pdf/yjbm00029-0044.pdf[↩][↩]
- Balwani M, Doheny D, Bishop DF, Nazarenko I, Yasuda M, Dailey HA, Anderson KE, Bissell DM, Bloomer J, Bonkovsky HL, Phillips JD, Liu L, Desnick RJ; Porphyrias Consortium of the National Institutes of Health Rare Diseases Clinical Research Network. Loss-of-function ferrochelatase and gain-of-function erythroid-specific 5-aminolevulinate synthase mutations causing erythropoietic protoporphyria and x-linked protoporphyria in North American patients reveal novel mutations and a high prevalence of X-linked protoporphyria. Mol Med. 2013 Apr 30;19(1):26-35. doi: 10.2119/molmed.2012.00340[↩][↩]
- Harms JH, Lautenschlager S, Minder CE, Minder EI. Mitigating photosensitivity of erythropoietic protoporphyria patients by an agonistic analog of alpha-melanocyte stimulating hormone. Photochem Photobiol. 2009 Nov-Dec;85(6):1434-9. doi: 10.1111/j.1751-1097.2009.00595.x[↩][↩]
- Minder EI. Afamelanotide, an agonistic analog of α-melanocyte-stimulating hormone, in dermal phototoxicity of erythropoietic protoporphyria. Expert Opin Investig Drugs. 2010 Dec;19(12):1591-602. doi: 10.1517/13543784.2010.535515[↩][↩]
- Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for Erythropoietic Protoporphyria. N Engl J Med. 2015 Jul 2;373(1):48-59. doi: 10.1056/NEJMoa1411481[↩][↩]
- Biolcati G, Marchesini E, Sorge F, Barbieri L, Schneider-Yin X, Minder EI. Long-term observational study of afamelanotide in 115 patients with erythropoietic protoporphyria. Br J Dermatol. 2015 Jun;172(6):1601-1612. doi: 10.1111/bjd.13598[↩][↩]
- Brancaleoni V, Balwani M, Granata F, Graziadei G, Missineo P, Fiorentino V, Fustinoni S, Cappellini MD, Naik H, Desnick RJ, Di Pierro E. X-chromosomal inactivation directly influences the phenotypic manifestation of X-linked protoporphyria. Clin Genet. 2016 Jan;89(1):20-6. doi: 10.1111/cge.12562[↩]
- Schneider-Yin X, Gouya L, Meier-Weinand A, Deybach JC, Minder EI. New insights into the pathogenesis of erythropoietic protoporphyria and their impact on patient care. Eur J Pediatr. 2000 Oct;159(10):719-25. doi: 10.1007/s004310000494[↩]
- Gou E, Weng C, Greene T, Anderson KE, Phillips JD. Longitudinal Analysis of Erythrocyte and Plasma Protoporphyrin Levels in Patients with Protoporphyria. J Appl Lab Med. 2018 Sep 1;3(2):213-221. doi: 10.1373/jalm.2017.025874[↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemias and the porphyrias. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2991-3062.[↩]
- Poh-Fitzpatrick MB. The “priming phenomenon” in the acute phototoxicity of erythropoietic protoporphyria. J Am Acad Dermatol. 1989 Aug;21(2 Pt 1):311. doi: 10.1016/s0190-9622(89)70187-0[↩][↩]
- Singal AK, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014 Sep;60(3):1082-9. doi: 10.1002/hep.27086[↩]
- Holme SA, Worwood M, Anstey AV, Elder GH, Badminton MN. Erythropoiesis and iron metabolism in dominant erythropoietic protoporphyria. Blood. 2007 Dec 1;110(12):4108-10. doi: 10.1182/blood-2007-04-088120[↩]
- Barman-Aksoezen J, Girelli D, Aurizi C, Schneider-Yin X, Campostrini N, Barbieri L, Minder EI, Biolcati G. Disturbed iron metabolism in erythropoietic protoporphyria and association of GDF15 and gender with disease severity. J Inherit Metab Dis. 2017 May;40(3):433-441. doi: 10.1007/s10545-017-0017-7[↩]
- Bossi K, Lee J, Schmeltzer P, Holburton E, Groseclose G, Besur S, Hwang S, Bonkovsky HL. Homeostasis of iron and hepcidin in erythropoietic protoporphyria. Eur J Clin Invest. 2015 Oct;45(10):1032-41. doi: 10.1111/eci.12503[↩]
- Holme SA, Anstey AV, Badminton MN, Elder GH. Serum 25-hydroxyvitamin D in erythropoietic protoporphyria. Br J Dermatol. 2008 Jul;159(1):211-3. doi: 10.1111/j.1365-2133.2008.08616.x[↩][↩]
- Spelt JM, de Rooij FW, Wilson JH, Zandbergen AA. Vitamin D deficiency in patients with erythropoietic protoporphyria. J Inherit Metab Dis. 2010 Dec;33 Suppl 3:S1-4. doi: 10.1007/s10545-008-1037-0[↩][↩]
- Biewenga M, Matawlie RHS, Friesema ECH, Koole-Lesuis H, Langeveld M, Wilson JHP, Langendonk JG. Osteoporosis in patients with erythropoietic protoporphyria. Br J Dermatol. 2017 Dec;177(6):1693-1698. doi: 10.1111/bjd.15893[↩]
- Gouya L, Puy H, Lamoril J, Da Silva V, Grandchamp B, Nordmann Y, Deybach JC. Inheritance in erythropoietic protoporphyria: a common wild-type ferrochelatase allelic variant with low expression accounts for clinical manifestation. Blood. 1999 Mar 15;93(6):2105-10. https://doi.org/10.1182/blood.V93.6.2105.406k28_2105_2110[↩]
- Gou EW, Balwani M, Bissell DM, Bloomer JR, Bonkovsky HL, Desnick RJ, Naik H, Phillips JD, Singal AK, Wang B, Keel S, Anderson KE. Pitfalls in Erythrocyte Protoporphyrin Measurement for Diagnosis and Monitoring of Protoporphyrias. Clin Chem. 2015 Dec;61(12):1453-6. doi: 10.1373/clinchem.2015.245456[↩]
- Wahlin S, Harper P. The role for BMT in erythropoietic protoporphyria. Bone Marrow Transplant. 2010 Feb;45(2):393-4. doi: 10.1038/bmt.2009.132[↩]
- Butler DF, Ginn KF, Daniel JF, Bloomer JR, Kats A, Shreve N, Myers GD. Bone marrow transplant for X-linked protoporphyria with severe hepatic fibrosis. Pediatr Transplant. 2015 Jun;19(4):E106-10. doi: 10.1111/petr.12472[↩]
- Wahlin S, Srikanthan N, Hamre B, Harper P, Brun A. Protection from phototoxic injury during surgery and endoscopy in erythropoietic protoporphyria. Liver Transpl. 2008 Sep;14(9):1340-6. doi: 10.1002/lt.21527[↩][↩][↩]
- Landefeld C, Kentouche K, Gruhn B, Stauch T, Rößler S, Schuppan D, Whatley SD, Beck JF, Stölzel U. X-linked protoporphyria: Iron supplementation improves protoporphyrin overload, liver damage and anaemia. Br J Haematol. 2016 May;173(3):482-4. doi: 10.1111/bjh.13612[↩]
- Balwani M. Effects of iron supplementation in EPP and XLP. Milan, Italy: International Congress on Porphyrins and Porphyria: From Bench to Care. 2019.[↩]
- Poh-Fitzpatrick MB. Human protoporphyria: reduced cutaneous photosensitivity and lower erythrocyte porphyrin levels during pregnancy. J Am Acad Dermatol. 1997 Jan;36(1):40-3. doi: 10.1016/s0190-9622(97)70323-2[↩]
- Study to Evaluate Efficacy, Safety, and Tolerability of MT-7117 in Subjects With Erythropoietic Protoporphyria. https://clinicaltrials.gov/study/NCT03520036[↩]
- Erwin A, Balwani M, Desnick RJ; Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network. Congenital Erythropoietic Porphyria. 2013 Sep 12 [Updated 2021 Apr 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK154652[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bhusal M, Bhattarai S, Shah M, Khadka A. Congenital erythropoietic porphyria: A case series of a rare uroporphyrinogen III synthase gene mutation in Nepalese patients. JAAD Case Rep. 2021 Feb 25;10:102-106. doi: 10.1016/j.jdcr.2021.02.017[↩]
- Enciso L. Hypertrichosis and erythrodontia in congenital erythropoietic porphyria. EJHaem. 2021 May 26;2(3):640-641. doi: 10.1002/jha2.215[↩]
- Goudet C, Ged C, Petit A, Desage C, Mahe P, Salhi A, Harzallah I, Blouin JM, Mercie P, Schmitt C, Poli A, Gouya L, Barlogis V, Richard E. Severe Perinatal Presentations of Günther’s Disease: Series of 20 Cases and Perspectives. Life (Basel). 2024 Jan 17;14(1):130. doi: 10.3390/life14010130[↩]
- Abkowitz JL. A simple Rx for congenital erythropoietic porphyria. Blood. 2020 Nov 19;136(21):2371-2372. doi: 10.1182/blood.2020007968[↩]
- Jia N, Yimin Y, Li M, Jiang L, Liu Y. Identification of a novel nonsense mutation and a recurrent missense mutation in UROS gene in a patient with congenital erythropoietic porphyria. Front Genet. 2025 Mar 31;16:1486595. doi: 10.3389/fgene.2025.1486595[↩]
- Bernardo-Seisdedos G, Charco JM, SanJuan I, García-Martínez S, Urquiza P, Eraña H, Castilla J, Millet O. Improving the Pharmacological Properties of Ciclopirox for Its Use in Congenital Erythropoietic Porphyria. J Pers Med. 2021 May 28;11(6):485. doi: 10.3390/jpm11060485[↩]
- Bragazzi Cunha J, Elenbaas JS, Maitra D, Kuo N, Azuero-Dajud R, Ferguson AC, Griffin MS, Lentz SI, Shavit JA, Omary MB. Acitretin mitigates uroporphyrin-induced bone defects in congenital erythropoietic porphyria models. Sci Rep. 2021 May 5;11(1):9601. doi: 10.1038/s41598-021-88668-9[↩]
- Khan J, Hashmi MU, Noor N, Khan AJ, Shrateh ON, Tahir MJ. Congenital erythropoietic porphyria presenting with recurrent epistaxis: a case report. J Med Case Rep. 2023 Nov 14;17(1):472. doi: 10.1186/s13256-023-04204-5[↩]
- Kahila A, Zamlout A, Mazloum A, Laila O, Badran A. Congenital erythropoietic porphyria (Gunther disease): a case report. Oxf Med Case Reports. 2020 Jul 24;2020(7):omaa051. doi: 10.1093/omcr/omaa051[↩][↩][↩]
- Sudrié-Arnaud B, Legendre M, Snanoudj S, Pelluard F, Bekri S, Tebani A. An Atypical Case of Congenital Erythropoietic Porphyria. Genes (Basel). 2021 Nov 19;12(11):1828. doi: 10.3390/genes12111828[↩]
- Gopalakrishna H, Mironova M, Malik S, Faust A, Khurram N, Koh C, Kleiner DE, Heller T. Porto-Sinusoidal Vascular Disease in Congenital Erythropoietic Porphyria Needing Liver Transplantation. ACG Case Rep J. 2024 Apr 26;11(5):e01336. doi: 10.14309/crj.0000000000001336[↩]
- Blouin JM, Ged C, Bernardo-Seisdedos G, Cabantous T, Pinson B, Poli A, Puy H, Millet O, Gouya L, Morice-Picard F, Richard E. Identification of novel UROS mutations in a patient with congenital erythropoietic porphyria and efficient treatment by phlebotomy. Mol Genet Metab Rep. 2021 Feb 11;27:100722. doi: 10.1016/j.ymgmr.2021.100722[↩]
- Saikrishna P, Palaniswamy G, Pillikunte Doddareddy N, Ishfaq L, Zargar MN, Wafa Eranhikkal F, Sahu S. Congenital Erythropoietic Porphyria: A Rare Inherited Disorder. Cureus. 2024 Mar 5;16(3):e55558. doi: 10.7759/cureus.55558[↩]
- Phillips JD, Steensma DP, Pulsipher MA, Spangrude GJ, Kushner JP. Congenital erythropoietic porphyria due to a mutation in GATA1: the first trans-acting mutation causative for a human porphyria. Blood. 2007 Mar 15;109(6):2618-21. doi: 10.1182/blood-2006-06-022848[↩][↩][↩][↩][↩][↩][↩][↩]
- Erwin AL, Desnick RJ. Congenital erythropoietic porphyria: Recent advances. Mol Genet Metab. 2019 Nov;128(3):288-297. doi: 10.1016/j.ymgme.2018.12.008[↩][↩][↩][↩][↩]
- Desjardins MP, Naccache L, Hébert A, Auger I, Teira P, Pelland-Marcotte MC. Very Early Diagnosis and Management of Congenital Erythropoietic Porphyria. Clin Pediatr (Phila). 2023 Jun;62(5):399-403. doi: 10.1177/00099228221128661[↩]
- Arora S, Harith AK, Sodhi N. Congenital Erythropoietic Porphyria with Undescended Testis. Indian J Dermatol. 2016 Jul-Aug;61(4):467. doi: 10.4103/0019-5154.185749[↩]
- Kamalyan M, Mohammadi M. Congenital erythropoietic porphyria five years observation with standard treatment: a case report. Oxf Med Case Reports. 2024 Jan 27;2024(1):omad151. doi: 10.1093/omcr/omad151[↩]
- Peterlin P, Bonnelye J, Garnier A, Le Bourgeois A, Guillaume T, Jullien M, Dutartre H, Le Moigne M, Schmitt C, Gouya L, Poli A, Barbarot S, Chevallier P. Successful treatment of congenital erythropoietic porphyria using matched unrelated hematopoietic stem cell transplantation in an adult: A case report. Skin Health Dis. 2024 Jan 27;4(2):e342. doi: 10.1002/ski2.342[↩]
- Chao CB, Zhang Y. Biochemistry, Uroporphyrinogen. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK546643[↩][↩][↩][↩][↩][↩]
- Desnick RJ, Astrin KH. Congenital erythropoietic porphyria: advances in pathogenesis and treatment. Br J Haematol. 2002 Jun;117(4):779-95. doi: 10.1046/j.1365-2141.2002.03557.x[↩][↩][↩][↩][↩][↩][↩][↩]
- Warner CA, Poh-Fitzpatrick MB, Zaider EF, Tsai SF, Desnick RJ. Congenital erythropoietic porphyria. A mild variant with low uroporphyrin I levels due to a missense mutation (A66V) encoding residual uroporphyrinogen III synthase activity. Arch Dermatol. 1992 Sep;128(9):1243-8. doi: 10.1001/archderm.128.9.1243[↩]
- R.P. Katugampola, A.V. Anstey, A.Y. Finlay, S. Whatley, J. Woolf, N. Mason, J.C. Deybach, H. Puy, C. Ged, H. de Verneuil, S. Hanneken, E. Minder, X. Schneider‐Yin, M.N. Badminton, A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases, British Journal of Dermatology, Volume 167, Issue 4, 1 October 2012, Pages 888–900, https://doi.org/10.1111/j.1365-2133.2012.11154.x[↩]
- Aizencang G, Solis C, Bishop DF, Warner C, Desnick RJ. Human uroporphyrinogen-III synthase: genomic organization, alternative promoters, and erythroid-specific expression. Genomics. 2000 Dec 1;70(2):223-31. doi: 10.1006/geno.2000.6373[↩][↩]
- Caiulo A, Nicolis S, Bianchi P, Zuffardi O, Bardoni B, Maraschio P, Ottolenghi S, Camerino G, Giglioni B. Mapping the gene encoding the human erythroid transcriptional factor NFE1-GF1 to Xp11.23. Hum Genet. 1991 Feb;86(4):388-90. doi: 10.1007/BF00201840[↩][↩]
- Di Pierro E, Russo R, Karakas Z, Brancaleoni V, Gambale A, Kurt I, Winter SS, Granata F, Czuchlewski DR, Langella C, Iolascon A, Cappellini MD. Congenital erythropoietic porphyria linked to GATA1-R216W mutation: challenges for diagnosis. Eur J Haematol. 2015 Jun;94(6):491-7. doi: 10.1111/ejh.12452[↩][↩][↩][↩][↩][↩][↩]
- Di Pierro E, Brancaleoni V, Granata F. Advances in understanding the pathogenesis of congenital erythropoietic porphyria. Br J Haematol. 2016 May;173(3):365-79. doi: 10.1111/bjh.13978[↩][↩][↩]
- Schroeder WA, Shelton JB, Shelton JR, Huynh V, Teplow DB. High performance liquid chromatographic separation of the globin chains of non-human hemoglobins. Hemoglobin. 1985;9:461–482. doi: 10.3109/03630268508997024[↩][↩]
- Bickers DR, Frank J, The Porphyrias. In: Fitzpatrick’s Dermatology in General Medicine, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Wolff K (eds), 8th (ed), McGraw-Hill, New York (NY), Chapter 132, (2012). https://accessmedicine.mhmedical.com/Content.aspx?bookId=392§ionId=41138853[↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias In: Metabolic The and Molecular Bases of Inherited Disease, 8th (ed), Scriver CR, Beaudet AL, Sly WS and Valle D, (eds), New York (NY), McGraw-Hill, (2014) 2961–3062.[↩][↩][↩][↩]
- Shirazi N., Chauhan B.P., Jindal R., Ahmad S. Congenital erythropoietic porphyria: a rare case of photosensitivity with hemolytic anaemia and mental retardation. J Coll Physicians Surg Pak. 2019;29(6):S23–S25. doi: 10.29271/jcpsp.2019.06.S23[↩]
- Congenital Erythropoietic Porphyria (CEP). https://porphyriafoundation.org/for-patients/types-of-porphyria/cep/[↩]
- Desnick RJ, Glass IA, Xu W, Solis C, Astrin KH. Molecular genetics of congenital erythropoietic porphyria. Semin Liver Dis. 1998;18(1):77-84. doi: 10.1055/s-2007-1007143[↩]
- Schulenburg-Brand D, Katugampola R, Anstey AV, Badminton MN. The cutaneous porphyrias. Dermatol Clin. 2014 Jul;32(3):369-84, ix. doi: 10.1016/j.det.2014.03.001[↩]
- Katugampola RP, Badminton MN, Finlay AY, Whatley S, Woolf J, Mason N, Deybach JC, Puy H, Ged C, de Verneuil H, Hanneken S, Minder E, Schneider-Yin X, Anstey AV. Congenital erythropoietic porphyria: a single-observer clinical study of 29 cases. Br J Dermatol. 2012 Oct;167(4):901-13. doi: 10.1111/j.1365-2133.2012.11160.x[↩][↩][↩]
- Warner CA, Yoo HW, Roberts AG, Desnick RJ. Congenital erythropoietic porphyria: identification and expression of exonic mutations in the uroporphyrinogen III synthase gene. J Clin Invest. 1992 Feb;89(2):693-700. https://pmc.ncbi.nlm.nih.gov/articles/instance/442904/pdf/jcinvest00046-0357.pdf[↩][↩]
- Poh-Fitzpatrick MB. The erythropoietic porphyrias. Dermatol Clin. 1986 Apr;4(2):291-6.[↩][↩]
- Fityan A, Fassihi H, Sarkany R. Congenital erythropoietic porphyria: mild presentation with late onset associated with a mutation in the UROS gene promoter sequence. Clin Exp Dermatol. 2016 Dec;41(8):953-954. doi: 10.1111/ced.12932[↩]
- Deybach JC, de Verneuil H, Phung N, Nordmann Y, Puissant A, Boffety B. Congenital erythropoietic porphyria (Günther’s disease): enzymatic studies on two cases of late onset. J Lab Clin Med. 1981 Apr;97(4):551-8.[↩]
- Murphy A, Gibson G, Elder GH, Otridge BA, Murphy GM. Adult-onset congenital erythropoietic porphyria (Günther’s disease) presenting with thrombocytopenia. J R Soc Med. 1995 Jun;88(6):357P-358P[↩]
- Pain RW, Welch FW, Woodroffe AJ, Handley DA, Lockwood WH. Erythropoietic uroporphyria of Gunther first presenting at 58 years with positive family studies. Br Med J. 1975 Sep 13;3(5984):621-3. doi: 10.1136/bmj.3.5984.621[↩][↩]
- Kramer S, Viljoen E, Meyer AM, Metz J. The anaemia of erythropoietic prophyria with the description of the disease in an elderly patient. Br J Haematol. 1965 Nov;11(6):666-75. doi: 10.1111/j.1365-2141.1965.tb00115.x[↩]
- Rao SU, Dar NR, Abbas M, Mumtaz J. Late onset erythropoietic porphyria (Gunther’s disease). J Coll Physicians Surg Pak. 2011 Sep;21(9):564-6.[↩]
- Fritsch C, Lang K, Bolsen K, Lehmann P, Ruzicka T. Congenital erythropoietic porphyria. Skin Pharmacol Appl Skin Physiol. 1998 Nov-Dec;11(6):347-57. doi: 10.1159/000029857[↩]
- Schmid R, Schwartz S, Sundberg RD, Erythropoietic (congenital) porphyria: A rare abnormality of the normoblasts. Blood 10 (1955) 416–428.[↩]
- Fritsch C, Bolsen K, Ruzicka T, Goerz G. Congenital erythropoietic porphyria. J Am Acad Dermatol. 1997 Apr;36(4):594-610. doi: 10.1016/s0190-9622(97)70249-4[↩][↩]
- Weston MJ, Nicholson DC, Lim CK, Clark KG, Macdonald A, Henderson MA, Williams R. Congenital erythropoietic uroporphyria (Günther’s disease) presenting in a middle aged man. Int J Biochem. 1978;9(12):921-6. doi: 10.1016/0020-711x(78)90071-x[↩]
- Venkatesh P, Garg SP, Kumaran E, Tewari HK. Congenital porphyria with necrotizing scleritis in a 9-year-old child. Clin Exp Ophthalmol. 2000 Aug;28(4):314-8. doi: 10.1046/j.1442-9071.2000.00330.x[↩]
- Siddique SS, Gonzalez-Gonzalez LA, Thakuria P, Chang PY, Foster CS. Scleral necrosis in a patient with congenital erythropoietic porphyria. Cornea. 2011 Jan;30(1):97-9. doi: 10.1097/ICO.0b013e3181e458fa[↩]
- Oguz F, Sidal M, Bayram C, Sansoy N, Hekim N. Ocular involvement in two symptomatic congenital erythropoietic porphyria. Eur J Pediatr. 1993 Aug;152(8):671-3. doi: 10.1007/BF01955245[↩]
- Piomelli S, Poh-Fitzpatrick MB, Seaman C, Skolnick LM, Berdon WE. Complete suppression of the symptoms of congenital erythropoietic porphyria by long-term treatment with high-level transfusions. N Engl J Med. 1986 Apr 17;314(16):1029-31. doi: 10.1056/NEJM198604173141607[↩][↩]
- Kontos AP, Ozog D, Bichakjian C, Lim HW. Congenital erythropoietic porphyria associated with myelodysplasia presenting in a 72-year-old man: report of a case and review of the literature. Br J Dermatol. 2003 Jan;148(1):160-4. doi: 10.1046/j.1365-2133.2003.05040.x[↩][↩]
- Laorr A, Greenspan A. Severe osteopenia in congenital erythropoietic porphyria. Can Assoc Radiol J. 1994 Aug;45(4):307-9.[↩]
- Daïkha-Dahmane F, Dommergues M, Narcy F, Gubler MC, Dumez Y, Gauthier E, Nordmann Y, Nessmann C, Terrasse G, Muller F. Congenital erythropoietic porphyria: prenatal diagnosis and autopsy findings in two sibling fetuses. Pediatr Dev Pathol. 2001 Mar-Apr;4(2):180-4. doi: 10.1007/s100240010143[↩]
- Tezcan I, Xu W, Gurgey A, Tuncer M, Cetin M, Oner C, Yetgin S, Ersoy F, Aizencang G, Astrin KH, Desnick RJ. Congenital erythropoietic porphyria successfully treated by allogeneic bone marrow transplantation. Blood. 1998 Dec 1;92(11):4053-8. https://doi.org/10.1182/blood.V92.11.4053[↩]
- Thomas C, Ged C, Nordmann Y, de Verneuil H, Pellier I, Fischer A, Blanche S. Correction of congenital erythropoietic porphyria by bone marrow transplantation. J Pediatr. 1996 Sep;129(3):453-6. doi: 10.1016/s0022-3476(96)70082-3[↩][↩]
- Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol. 2001 Aug;45(2):279-82. doi: 10.1067/mjd.2001.114730[↩]
- Shaw PH, Mancini AJ, McConnell JP, Brown D, Kletzel M. Treatment of congenital erythropoietic porphyria in children by allogeneic stem cell transplantation: a case report and review of the literature. Bone Marrow Transplant. 2001 Jan;27(1):101-5. doi: 10.1038/sj.bmt.1702738[↩]
- Dupuis-Girod S, Akkari V, Ged C, Galambrun C, Kebaïli K, Deybach JC, Claudy A, Geburher L, Philippe N, de Verneuil H, Bertrand Y. Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Günther disease). Eur J Pediatr. 2005 Feb;164(2):104-7. doi: 10.1007/s00431-004-1575-x[↩]
- Taibjee SM, Stevenson OE, Abdullah A, Tan CY, Darbyshire P, Moss C, Goodyear H, Heagerty A, Whatley S, Badminton MN. Allogeneic bone marrow transplantation in a 7-year-old girl with congenital erythropoietic porphyria: a treatment dilemma. Br J Dermatol. 2007 Mar;156(3):567-71. doi: 10.1111/j.1365-2133.2006.07699.x[↩]
- Faraci M, Morreale G, Boeri E, Lanino E, Dallorso S, Dini G, Scuderi F, Cohen A, Cappelli B. Unrelated HSCT in an adolescent affected by congenital erythropoietic porphyria. Pediatr Transplant. 2008 Feb;12(1):117-20. doi: 10.1111/j.1399-3046.2007.00842.x[↩]
- Katugampola RP, Anstey AV, Finlay AY, Whatley S, Woolf J, Mason N, Deybach JC, Puy H, Ged C, de Verneuil H, Hanneken S, Minder E, Schneider-Yin X, Badminton MN. A management algorithm for congenital erythropoietic porphyria derived from a study of 29 cases. Br J Dermatol. 2012 Oct;167(4):888-900. doi: 10.1111/j.1365-2133.2012.11154.x[↩][↩][↩]
- Zix-Kieffer I, Langer B, Eyer D, Acar G, Racadot E, Schlaeder G, Oberlin F, Lutz P. Successful cord blood stem cell transplantation for congenital erythropoietic porphyria (Gunther’s disease). Bone Marrow Transplant. 1996 Jul;18(1):217-20.[↩][↩]
- Kauffman L, Evans DI, Stevens RF, Weinkove C. Bone-marrow transplantation for congenital erythropoietic porphyria. Lancet. 1991 Jun 22;337(8756):1510-1. doi: 10.1016/0140-6736(91)93198-i[↩]
- Hallai N, Anstey A, Mendelsohn S, Williams J, Evans-Jones G, Malick S, Badminton MN. Pregnancy in a patient with congenital erythropoietic porphyria. N Engl J Med. 2007 Aug 9;357(6):622-3. doi: 10.1056/NEJMc070009[↩]
- Sarkany RP, Ibbotson SH, Whatley SD, Lawrence CM, Gover P, Mufti GJ, Murphy GM, Masters GS, Badminton MN, Elder GH. Erythropoietic uroporphyria associated with myeloid malignancy is likely distinct from autosomal recessive congenital erythropoietic porphyria. J Invest Dermatol. 2011 May;131(5):1172-5. doi: 10.1038/jid.2011.5[↩]
- Cernik C, Haller N, Mostow EN. Adult-onset erythropoietic porphyria in the setting of MDS. Arch Dermatol. 2009 Aug;145(8):948-9. doi: 10.1001/archdermatol.2009.161[↩]
- Aguilera P, Badenas C, Whatley SD, To-Figueras J. Late-onset cutaneous porphyria in a patient heterozygous for a uroporphyrinogen III synthase gene mutation. Br J Dermatol. 2016 Dec;175(6):1346-1350. doi: 10.1111/bjd.14675[↩]
- Gonzalez-Mosquera LF, Sonthalia S. Acute Intermittent Porphyria. [Updated 2023 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK547665[↩][↩][↩][↩][↩][↩][↩][↩]
- Sardh E, Barbaro M. Acute Intermittent Porphyria. 2005 Sep 27 [Updated 2024 Feb 8]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1193[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Bustad HJ, Kallio JP, Vorland M, Fiorentino V, Sandberg S, Schmitt C, Aarsand AK, Martinez A. Acute Intermittent Porphyria: An Overview of Therapy Developments and Future Perspectives Focusing on Stabilisation of HMBS and Proteostasis Regulators. Int J Mol Sci. 2021 Jan 12;22(2):675. doi: 10.3390/ijms22020675[↩]
- Lei JJ, Li S, Dong BX, Yang J, Ren Y. Acute intermittent porphyria: a disease with low penetrance and high heterogeneity. Front Genet. 2024 Aug 12;15:1374965. doi: 10.3389/fgene.2024.1374965[↩][↩]
- Ren Y, Li S, Lei JJ, Li R, Dong BX, Yang J. Clinical feature and genetic analysis of HMBS gene in Chinese patients with acute intermittent porphyria: a systematic review. Front Genet. 2023 Dec 11;14:1291719. doi: 10.3389/fgene.2023.1291719[↩]
- Longo M, Paolini E, Meroni M, Dongiovanni P. Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution. Biomedicines. 2022 Mar 11;10(3):648. doi: 10.3390/biomedicines10030648[↩]
- Patel P, Midha S, Shukla S, Dhamija D, Bello AO, Khan S. Evaluating the Efficacy of a Small Interfering Ribonucleic Acid Molecule, Givosiran, in Treating Acute Intermittent Porphyria: A Systematic Review. Cureus. 2023 Jun 18;15(6):e40585. doi: 10.7759/cureus.40585[↩]
- Li S, Lei JJ, Dong BX, Ren Y, Yang J. HMBS gene mutations and hydroxymethylbilane synthase activity in acute intermittent porphyria: A systematic review. Medicine (Baltimore). 2023 Sep 29;102(39):e35144. doi: 10.1097/MD.0000000000035144[↩][↩]
- Storjord E, Wahlin S, Karlsen BO, Hardersen RI, Dickey AK, Ludviksen JK, Brekke OL. Potential Biomarkers for the Earlier Diagnosis of Kidney and Liver Damage in Acute Intermittent Porphyria. Life (Basel). 2023 Dec 21;14(1):19. doi: 10.3390/life14010019[↩]
- Acute Intermittent Porphyria (AIP). https://porphyriafoundation.org/for-patients/types-of-porphyria/aip/[↩]
- Acute Intermittent Porphyria. https://rarediseases.org/rare-diseases/acute-intermittent-porphyria/[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Founder Effect. https://www.genome.gov/genetics-glossary/Founder-Effect[↩]
- Cederlöf M, Bergen SE, Larsson H, Landén M, Lichtenstein P. Acute intermittent porphyria: comorbidity and shared familial risks with schizophrenia and bipolar disorder in Sweden. Br J Psychiatry. 2015 Dec;207(6):556-7. doi: 10.1192/bjp.bp.114.157073[↩][↩]
- Gouya L, Ventura P, Balwani M,, et al. EXPLORE: A Prospective, Multinational, Natural History Study of Patients with Acute Hepatic Porphyria with Recurrent Attacks. Hepatology. 2020 May;71(5):1546-1558. doi: 10.1002/hep.30936[↩][↩][↩]
- Acute Intermittent Porphyria. https://emedicine.medscape.com/article/205220-overview[↩][↩][↩]
- Schulenburg-Brand D., Stewart F., Stein P., Rees D., Badminton M. (2022). Update on the diagnosis and management of the autosomal dominant acute hepatic porphyrias. J. Clin. Pathol. 75, 537–543. 10.1136/jclinpath-2021-207647[↩][↩]
- Wang B., Bonkovsky H. L., Lim J. K., Balwani M. (2023). AGA clinical practice update on diagnosis and management of acute hepatic porphyrias: expert review. Gastroenterology 164, 484–491. 10.1053/j.gastro.2022.11.034[↩]
- Yarra P., Faust D., Bennett M., Rudnick S., Bonkovsky H. L. (2019). Benefits of prophylactic heme therapy in severe acute intermittent porphyria. Mol. Genet. Metab. Rep. 19, 100450. 10.1016/j.ymgmr.2019.01.002[↩]
- Blaylock B., Epstein J., Stickler P. (2020). Real-world annualized healthcare utilization and expenditures among insured US patients with acute intermittent porphyria (AIP) treated with hemin. J. Med. Econ. 23, 537–545. 10.1080/13696998.2020.1724118[↩]
- Poli A, Schmitt C, Moulouel B, et al. Givosiran in acute intermittent porphyria: A personalized medicine approach. Mol Genet Metab. 2022 Mar;135(3):206-214. doi: 10.1016/j.ymgme.2022.01.002[↩]
- Kizilaslan EZ, Ghadge NM, Martinez A, Bass M, Winayak R, Mathew M, Amin R, Khan M, Kizilbash N. Acute Intermittent Porphyria’s Symptoms and Management: A Narrative Review. Cureus. 2023 Mar 13;15(3):e36058. doi: 10.7759/cureus.36058[↩]
- Kizilaslan EZ, Ghadge NM, Martinez A, Bass M, Winayak R, Mathew M, Amin R, Khan M, Kizilbash N. Acute Intermittent Porphyria’s Symptoms and Management: A Narrative Review. Cureus. 2023 Mar 13;15(3):e36058. doi: 10.7759/cureus.36058[↩]
- Namba H, Narahara K, Tsuji K, Yokoyama Y, Seino Y. Assignment of human porphobilinogen deaminase to 11q24.1—-q24.2 by in situ hybridization and gene dosage studies. Cytogenet Cell Genet. 1991;57(2-3):105-8. doi: 10.1159/000133123[↩]
- Szlendak U, Bykowska K, Lipniacka A. Clinical, Biochemical and Molecular Characteristics of the Main Types of Porphyria. Adv Clin Exp Med. 2016 Mar-Apr;25(2):361-8. doi: 10.17219/acem/58955[↩]
- HMBS gene. https://medlineplus.gov/genetics/gene/hmbs/[↩]
- Anderson K. E., Bloomer J. R., Bonkovsky H. L., Kushner J. P., Pierach C. A., Pimstone N. R., et al. (2005). Recommendations for the diagnosis and treatment of the acute porphyrias. Ann. Intern Med. 142, 439–450. 10.7326/0003-4819-142-6-200503150-00010[↩]
- Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010 Mar 13;375(9718):924-37. doi: 10.1016/S0140-6736(09)61925-5[↩][↩]
- Chen B, Solis-Villa C, Hakenberg J, Qiao W, Srinivasan RR, Yasuda M, Balwani M, Doheny D, Peter I, Chen R, Desnick RJ. Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease. Hum Mutat. 2016 Nov;37(11):1215-1222. doi: 10.1002/humu.23067[↩]
- Kuo HC, Huang CC, Chu CC, Lee MJ, Chuang WL, Wu CL, Wu T, Ning HC, Liu CY. Neurological complications of acute intermittent porphyria. Eur Neurol. 2011;66(5):247-52. doi: 10.1159/000330683[↩]
- Storjord E, Dahl JA, Landsem A, Fure H, Ludviksen JK, Goldbeck-Wood S, Karlsen BO, Berg KS, Mollnes TE, W Nielsen E, Brekke OL. Systemic inflammation in acute intermittent porphyria: a case-control study. Clin Exp Immunol. 2017 Mar;187(3):466-479. doi: 10.1111/cei.12899[↩][↩]
- Barreda-Sánchez M., Buendía-Martínez J., Glover-López G., Carazo-Díaz C., Ballesta-Martínez M. J., López-González V., et al. (2019). High penetrance of acute intermittent porphyria in a Spanish founder mutation population and CYP2D6 genotype as a susceptibility factor. Orphanet J. Rare Dis. 14, 59. 10.1186/s13023-019-1031-7[↩]
- Wang B, Bonkovsky HL, Lim JK, Balwani M. AGA Clinical Practice Update on Diagnosis and Management of Acute Hepatic Porphyrias: Expert Review. Gastroenterology. 2023 Mar;164(3):484-491. doi: 10.1053/j.gastro.2022.11.034[↩]
- Stein PE, Edel Y, Mansour R, Mustafa RA, Sandberg S; Members of the Acute Porphyria Expert Panel. Key terms and definitions in acute porphyrias: Results of an international Delphi consensus led by the European porphyria network. J Inherit Metab Dis. 2023 Jul;46(4):662-674. doi: 10.1002/jimd.12612[↩][↩][↩][↩]
- Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol Commun. 2018 Dec 20;3(2):193-206. doi: 10.1002/hep4.1297[↩][↩][↩][↩][↩]
- Vassiliou D, Sardh E. Acute hepatic porphyria and maternal health: Clinical and biochemical follow-up of 44 pregnancies. J Intern Med. 2022 Jan;291(1):81-94. doi: 10.1111/joim.13376[↩][↩]
- Mantel Ä, Vassiliou D, Lissing M, Stephansson O, Wahlin S, Sardh E. Maternal and fetal outcomes in acute hepatic porphyria: A Swedish National Cohort Study. J Inherit Metab Dis. 2023 Jul;46(4):675-686. doi: 10.1002/jimd.12616. Epub 2023 Apr 26. Erratum in: J Inherit Metab Dis. 2024 Jan;47(1):212. doi: 10.1002/jimd.12685[↩][↩][↩]
- Storjord E, Dahl JA, Landsem A, Ludviksen JK, Karlsen MB, Karlsen BO, Brekke OL. Lifestyle factors including diet and biochemical biomarkers in acute intermittent porphyria: Results from a case-control study in northern Norway. Mol Genet Metab. 2019 Nov;128(3):254-270. doi: 10.1016/j.ymgme.2018.12.006[↩]
- Pischik E, Baumann K, Karpenko A, Kauppinen R. Pathogenesis of acute encephalopathy in acute hepatic porphyria. J Neurol. 2023 May;270(5):2613-2630. doi: 10.1007/s00415-023-11586-5[↩][↩][↩][↩]
- Sardh E, Harper P. RNAi therapy with givosiran significantly reduces attack rates in acute intermittent porphyria. J Intern Med. 2022 May;291(5):593-610. doi: 10.1111/joim.13443[↩][↩][↩]
- Marsden JT, Rees DC. Urinary excretion of porphyrins, porphobilinogen and δ-aminolaevulinic acid following an attack of acute intermittent porphyria. J Clin Pathol. 2014 Jan;67(1):60-5. doi: 10.1136/jclinpath-2012-201367[↩]
- Lissing M, Vassiliou D, Floderus Y, Harper P, Bottai M, Kotopouli M, Hagström H, Sardh E, Wahlin S. Risk of primary liver cancer in acute hepatic porphyria patients: A matched cohort study of 1244 individuals. J Intern Med. 2022 Jun;291(6):824-836. doi: 10.1111/joim.13463[↩]
- Baumann K, Kauppinen R. Penetrance and predictive value of genetic screening in acute porphyria. Mol Genet Metab. 2020 May;130(1):87-99. doi: 10.1016/j.ymgme.2020.02.003[↩]
- Fontanellas A, Ávila MA, Berraondo P. Emerging therapies for acute intermittent porphyria. Expert Rev Mol Med. 2016 Nov 2;18:e17. doi: 10.1017/erm.2016.18[↩]
- O’Malley R, Rao G, Stein P, Bandmann O. Porphyria: often discussed but too often missed. Pract Neurol. 2018 Oct;18(5):352-358. doi: 10.1136/practneurol-2017-001878[↩]
- Simon A, Pompilus F, Querbes W, Wei A, Strzok S, Penz C, Howe DL, Hungate JR, Kim JB, Agarwal S, Marquis P. Patient Perspective on Acute Intermittent Porphyria with Frequent Attacks: A Disease with Intermittent and Chronic Manifestations. Patient. 2018 Oct;11(5):527-537. doi: 10.1007/s40271-018-0319-3[↩]
- Pallet N, Mami I, Schmitt C, Karim Z, François A, Rabant M, Nochy D, Gouya L, Deybach JC, Xu-Dubois Y, Thervet E, Puy H, Karras A. High prevalence of and potential mechanisms for chronic kidney disease in patients with acute intermittent porphyria. Kidney Int. 2015 Aug;88(2):386-95. doi: 10.1038/ki.2015.97[↩]
- Sardh E, Andersson DE, Henrichson A, Harper P. Porphyrin precursors and porphyrins in three patients with acute intermittent porphyria and end-stage renal disease under different therapy regimes. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):66-71.[↩]
- Ramanujam VS, Anderson KE. Porphyria Diagnostics-Part 1: A Brief Overview of the Porphyrias. Curr Protoc Hum Genet. 2015 Jul 1;86:17.20.1-17.20.26. doi: 10.1002/0471142905.hg1720s86[↩]
- Besur S, Schmeltzer P, Bonkovsky HL. Acute Porphyrias. J Emerg Med. 2015 Sep;49(3):305-12. doi: 10.1016/j.jemermed.2015.04.034[↩][↩]
- Stewart MF. Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow-up. J Clin Pathol. 2012 Nov;65(11):976-80. doi: 10.1136/jclinpath-2012-200791[↩]
- Bissell DM, Anderson KE, Bonkovsky HL. Porphyria. N Engl J Med. 2017 Nov 23;377(21):2101. doi: 10.1056/NEJMc1712682[↩]
- Stein PE, Badminton MN, Rees DC. Update review of the acute porphyrias. Br J Haematol. 2017 Feb;176(4):527-538. doi: 10.1111/bjh.14459[↩][↩]
- Nguyen TP, Taylor RS. Guillain-Barre Syndrome. [Updated 2023 Feb 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532254[↩]
- Hadden RD, Cornblath DR, Hughes RA, Zielasek J, Hartung HP, Toyka KV, Swan AV. Electrophysiological classification of Guillain-Barré syndrome: clinical associations and outcome. Plasma Exchange/Sandoglobulin Guillain-Barré Syndrome Trial Group. Ann Neurol. 1998 Nov;44(5):780-8. doi: 10.1002/ana.410440512[↩]
- Rocha Cabrero F, Morrison EH. Miller Fisher Syndrome. [Updated 2023 Jun 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507717[↩]
- McKhann GM, Cornblath DR, Griffin JW, Ho TW, Li CY, Jiang Z, Wu HS, Zhaori G, Liu Y, Jou LP, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993 Apr;33(4):333-42. doi: 10.1002/ana.410330402[↩]
- Hafer-Macko C, Hsieh ST, Li CY, Ho TW, Sheikh K, Cornblath DR, McKhann GM, Asbury AK, Griffin JW. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. 1996 Oct;40(4):635-44. doi: 10.1002/ana.410400414[↩]
- Roveri G, Nascimbeni F, Rocchi E, Ventura P. Drugs and acute porphyrias: reasons for a hazardous relationship. Postgrad Med. 2014 Nov;126(7):108-20. doi: 10.3810/pgm.2014.11.2839[↩][↩]
- Lissing M, Nowak G, Adam R, Karam V, Boyd A, Gouya L, Meersseman W, Melum E, Ołdakowska-Jedynak U, Reiter FP, Colmenero J, Sanchez R, Herden U, Langendonk J, Ventura P, Isoniemi H, Boillot O, Braun F, Perrodin S, Mowlem E, Wahlin S; European Liver and Intestine Transplant Association. Liver Transplantation for Acute Intermittent Porphyria. Liver Transpl. 2021 Apr;27(4):491-501. doi: 10.1002/lt.25959[↩][↩][↩]
- Dowman JK, Gunson BK, Bramhall S, Badminton MN, Newsome PN. Liver transplantation from donors with acute intermittent porphyria. Ann Intern Med. 2011 Apr 19;154(8):571-2. doi: 10.7326/0003-4819-154-8-201104190-00015. Erratum in: Ann Intern Med. 2011 Apr 19;154(8):572. Erratum in: Ann Intern Med. 2011 Jun 21;154(12):848.[↩]
- Pallet N, Karras A, Thervet E, Gouya L, Karim Z, Puy H. Porphyria and kidney diseases. Clin Kidney J. 2018 Apr;11(2):191-197. doi: 10.1093/ckj/sfx146[↩]
- Wahlin S, Harper P, Sardh E, Andersson C, Andersson DE, Ericzon BG. Combined liver and kidney transplantation in acute intermittent porphyria. Transpl Int. 2010 Jun;23(6):e18-21. doi: 10.1111/j.1432-2277.2009.01035.x[↩]
- Schulenburg-Brand D, Gardiner T, Guppy S, Rees DC, Stein P, Barth J, Felicity Stewart M, Badminton M. An Audit of the Use of Gonadorelin Analogues to Prevent Recurrent Acute Symptoms in Patients with Acute Porphyria in the United Kingdom. JIMD Rep. 2017;36:99-107. doi: 10.1007/8904_2017_2[↩]
- Johansson A, Möller C, Fogh J, Harper P. Biochemical characterization of porphobilinogen deaminase-deficient mice during phenobarbital induction of heme synthesis and the effect of enzyme replacement. Mol Med. 2003 Sep-Dec;9(9-12):193-9. doi: 10.2119/2004-00002.johansson[↩]
- Sardh E, Rejkjaer L, Andersson DE, Harper P. Safety, pharmacokinetics and pharmocodynamics of recombinant human porphobilinogen deaminase in healthy subjects and asymptomatic carriers of the acute intermittent porphyria gene who have increased porphyrin precursor excretion. Clin Pharmacokinet. 2007;46(4):335-49. doi: 10.2165/00003088-200746040-00006[↩]
- Prieto J, Gonzalez-Aseguinolaza G. Acute Intermittent Porphyria: Novel Etiologic and Pathogenic Therapies Based on RNA Transfer to the Liver. Hepatology. 2019 Sep;70(3):1061-1063. doi: 10.1002/hep.30678[↩]
- Vassiliou D, Lempessi C, Harper P, Sardh E. Challenges in the management of acute intermittent porphyria with recurrent attacks during pregnancy: A case report. Clin Case Rep. 2020 Jul 30;8(12):2483-2487. doi: 10.1002/ccr3.3185[↩]
- Pischik E, Kauppinen R. An update of clinical management of acute intermittent porphyria. Appl Clin Genet. 2015 Sep 1;8:201-14. doi: 10.2147/TACG.S48605[↩]
- Lissing M, Vassiliou D, Floderus Y, Harper P, Yan J, Hagström H, Sardh E, Wahlin S. Risk for incident comorbidities, nonhepatic cancer and mortality in acute hepatic porphyria: A matched cohort study in 1244 individuals. J Inherit Metab Dis. 2023 Mar;46(2):286-299. doi: 10.1002/jimd.12583[↩]
- Kauppinen R, Mustajoki P. Prognosis of acute porphyria: occurrence of acute attacks, precipitating factors, and associated diseases. Medicine (Baltimore). 1992 Jan;71(1):1-13.[↩]
- Wang B, Bissell DM. Hereditary Coproporphyria. 2012 Dec 13 [Updated 2022 May 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK114807[↩][↩][↩][↩][↩][↩]
- Towns C, Balakrishnan S, Florkowski C, Davies A, Barrington-Ward E. High penetrance, recurrent attacks and thrombus formation in a family with hereditary coproporphyria. JIMD Rep. 2022 Mar 18;63(3):211-215. doi: 10.1002/jmd2.12281[↩]
- Kaftory R, Edel Y, Snast I, Lapidoth M, Mamet R, Elis A, Hodak E, Levi A. Greater disease burden of variegate porphyria than hereditary coproporphyria: An Israeli nationwide study of neurocutaneous porphyrias. Mol Genet Metab Rep. 2021 Jan 13;26:100707. doi: 10.1016/j.ymgmr.2021.100707[↩]
- Eroglu S, Birsenogul I. Delirium with delayed diagnosis of hereditary coproporphyria. Clin Case Rep. 2022 Jun 2;10(6):e05937. doi: 10.1002/ccr3.5937[↩]
- Edel Y, Mamet R, Sagy I, Snast I, Kaftory R, Mimouni T, Levi A. A 25-Hour Fast Among Quiescent Hereditary Coproporphyria and Variegate Porphyria Patients is Associated With a Low Risk of Complications. Rambam Maimonides Med J. 2023 Jan 29;14(1):e0003. doi: 10.5041/RMMJ.10490[↩]
- Whatley SD, Mason NG, Woolf JR, Newcombe RG, Elder GH, Badminton MN. Diagnostic strategies for autosomal dominant acute porphyrias: retrospective analysis of 467 unrelated patients referred for mutational analysis of the HMBS, CPOX, or PPOX gene. Clin Chem. 2009 Jul;55(7):1406-14. doi: 10.1373/clinchem.2008.122564[↩]
- Anderson KE, Bloomer JR, Bonkovsky HL, Kushner JP, Pierach CA, Pimstone NR, Desnick RJ. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005 Mar 15;142(6):439-50. doi: 10.7326/0003-4819-142-6-200503150-00010. Erratum in: Ann Intern Med. 2005 Aug 16;143(4):316.[↩]
- Valle, G. , Carmine Guida, C. , Nasuto, M. , Totaro, M. , Aucella, F. , Frusciante, V. , Di Mauro, L. , Potenza, A. , Savino, M. , Stanislao, M. , Popolizio, T. , Guglielmi, G. , Giagulli, V. A. , Guastamacchia, E. , & Triggiani, V. (2016). Cerebral hypoperfusion in hereditary coproporphyria (HCP): A single photon emission computed tomography (SPECT) study. Endocrine, Metabolic & Immune Disorders‐Drug Targets, 16, 39–46. 10.2174/1871530316666151218151101[↩]
- Liu A, Zhou L, Zhu H, Li Y, Yang J. Systemic Lupus Erythematosus and Hereditary Coproporphyria: Two Different Entities Diagnosed by WES in the Same Patient. Biomed Res Int. 2022 May 28;2022:9096999. doi: 10.1155/2022/9096999[↩]
- Hereditary coproporphyria. https://rarediseases.org/rare-diseases/hereditary-coproporphyria[↩][↩]
- Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute Hepatic Porphyrias: Review and Recent Progress. Hepatol Commun. 2018;3(2):193–206. Published 2018 Dec 20. doi:10.1002/hep4.1297 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6357830[↩]
- Balwani M, Wang B, Anderson KE, Bloomer JR, Bissell DM, Bonkovsky HL, Phillips JD, Desnick RJ, et al.. Acute hepatic porphyrias: recommendations for evaluation and long-term management. Hepatology. 2017; 66:1314-1322. https://www.ncbi.nlm.nih.gov/pubmed/28605040[↩][↩]
- Ma E, Mar V, Varigos G, Nicoll A, Ross G. Haem arginate as effective maintenance therapy for hereditary coproporphyria. Australas J Dermatol. 2011 May;52(2):135-8. doi: 10.1111/j.1440-0960.2011.00747.x[↩][↩]
- Kühnel A, Gross U, Doss MO. Hereditary coproporphyria in Germany: clinical-biochemical studies in 53 patients. Clin Biochem. 2000 Aug;33(6):465-73. doi: 10.1016/s0009-9120(00)00159-4[↩]
- Stein PE, Badminton MN, Rees DC.. Update review of the acute porphyrias. Br Jl Hemot. 2017; 176(4):527-538. https://www.ncbi.nlm.nih.gov/pubmed/27982422[↩]
- Balwani M, Sardh E, Ventura P, Peiró PA, Rees DC, et al. ENVISION Investigators. Phase 3 Trial of RNAi Therapeutic Givosiran for Acute Intermittent Porphyria. N Engl J Med. 2020 Jun 11;382(24):2289-2301. doi: 10.1056/NEJMoa1913147[↩]
- Balwani M, Naik H, Anderson KE, et al. Clinical, biochemical, and genetic characterization of North American patients with erythropoietic protoporphyria and x-linked protoporphyria. JAMA Dermatology. 2017;153(8):789–796. doi: 10.1001/jamadermatol.2017.1557[↩]
- Wang B, Rudnick S, Cengia B, Bonkovsky HL. Acute hepatic porphyrias: review and recent progress. Hepatology Communications. 2018;3(2):193–206. doi: 10.1002/hep4.1297[↩][↩]





{kind=link}