Contents
What is Stickler syndrome
Stickler syndrome is a group of hereditary multisystem connective tissue disorder characterized by a distinctive facial appearance (mid-facial flatness, small chin, long upper lip or philtrum), palatal abnormalities (cleft palate, bifid uvula or high arched palate), eye abnormalities (vitreoretinal degeneration, myopia, cataracts, retinal holes and retinal detachments), hearing loss (sensorineural hearing loss), skeleton and joint problems (loose joints, scoliosis, chest deformities, a hip disorder of childhood [Legg-Calve-Perthe’s disease], early onset degenerative osteoarthritis before age 40 years) and mitral valve prolapse 1, 2, 3, 4, 5, 6, 7, 8, 9. These signs and symptoms present in Stickler syndrome often vary greatly from one individual to another. Stickler syndrome signs and symptoms can include eye findings of myopia, cataract, and retinal detachment (50% life-time risk of retinal detachment with minor eye trauma causing vitreous hemorrhage and/or retinal detachments); hearing loss that is both conductive and sensorineural; midfacial underdevelopment and cleft palate (either alone or as part of the Robin sequence); and mild spondyloepiphyseal dysplasia and/or precocious arthritis.
Stickler syndrome received its name from Dr. Gunnar B. Stickler, who first studied and documented the syndrome at Mayo Clinic in 1965 6. A syndrome is a collection of specific symptoms, all with one cause. Dr. Stickler called the disorder hereditary progressive arthro-ophthalmopathy. Connective tissue is the material between cells of the body that gives the tissue form and strength and is found all over the body. Connective tissue is made up of a protein known as collagen of which there are several different varieties found in the body. Stickler syndrome often affects the connective tissue of the eye, especially in the interior of the eyeball (vitreous humor), the specialized tissue that serves as a buffer or cushion for bones at joints (cartilage) and the ends of the bones that make up the joints of the body (epiphysis).
A characteristic feature of Stickler syndrome is a somewhat flattened facial appearance. This appearance results from underdeveloped bones in the middle of the face, including the cheekbones and the bridge of the nose. A particular group of physical features called Pierre Robin sequence is also common in people with Stickler syndrome. Pierre Robin sequence includes an opening in the roof of the mouth (a cleft palate), a tongue that is placed further back than normal (glossoptosis), and a small lower jaw (micrognathia). This combination of features can lead to feeding problems and difficulty breathing. Approximately 30% to 40% of patients with Pierre Robin sequence have Stickler syndrome 10, 11.
Many people with Stickler syndrome have severe nearsightedness (high myopia). In some cases, the clear gel that fills the eyeball (the vitreous) has an abnormal appearance, which is noticeable during an eye examination. Other eye problems are also common, including increased pressure within the eye (glaucoma), clouding of the lens of the eyes (cataracts), and tearing of the lining of the eye (retinal detachment). These eye abnormalities cause impaired vision or blindness in some cases.
In people with Stickler syndrome, hearing loss varies in degree and may become more severe over time. The hearing loss may be sensorineural, meaning that it results from changes in the inner ear, or conductive, meaning that it is caused by abnormalities of the middle ear.
Most people with Stickler syndrome have skeletal abnormalities that affect the joints. The joints of affected children and young adults may be loose and very flexible (hypermobile), though joints become less flexible with age. Arthritis often appears early in life and may cause joint pain or stiffness. Problems with the bones of the spine (vertebrae) can also occur, including abnormal curvature of the spine (scoliosis or kyphosis) and flattened vertebrae (platyspondyly). These spinal abnormalities may cause back pain.
Researchers have described several types of Stickler syndrome, which are distinguished by their genetic causes and their patterns of signs and symptoms. In particular, the eye abnormalities and severity of hearing loss differ among the types. Type I has the highest risk of retinal detachment. Type II also includes eye abnormalities, but type III does not (and is often called non-ocular Stickler syndrome). Types II and III are more likely than type I to have significant hearing loss. Types IV, V, and VI are very rare and have each been diagnosed in only a few individuals. Stickler syndrome affects an estimated 1 in 7,500 to 9,000 newborns 7, 8.
Table 1. Stickler Syndrome associated genes and its associated physical features
| Physical Feature | % of Persons with Physical Feature 1 | |||
|---|---|---|---|---|
| COL2A1 Stickler syndrome | COL11A1 Stickler syndrome | COL11A2 Stickler syndrome | COL9A1-, COL9A2-, & COL9A3 Stickler syndrome | |
| Cleft palate | 30%-60% | 60% | 35% | — |
| Myopia | 80%-90% | 80%-85% | — | 90%-95% |
| Retinal detachment | 40%-70% | <40% | — | 13%-18% |
| Hearing loss | 20%-50% (sensorineural hearing loss ± conductive hearing loss) | 75%-80% | 60% | 90%-95% (sensorineural hearing loss) |
| Skeletal signs and symptoms 2 | 35%-40% | 25% | 50% | 25% |
Footnotes: — = not reported; HL = hearing loss; SNHL = sensorineural hearing loss; SS = Stickler syndrome
2 Includes mainly early-onset degenerative joint disease
[Source 14 ]Stickler syndrome type I (STL1) is responsible for approximately 80% to 90% of reported Stickler syndrome cases (most common form of Stickler syndrome) and presents with a wide variety of symptoms affecting the eye, ear, facial appearance, palate and musculoskeletal system 15, 12. Most will have ‘full’ Stickler syndrome affecting the sight, joints, hearing and any mid-line clefting. Findings show those with this anomaly have an increased incidence of cleft abnormalities. Stickler syndrome type I (STL1) is caused to mutations over the entire COL2A1 gene on chromosome 12q13.11 16, 17, 18. These mutations cause loss of function of the COL2A1 gene. The majority of these mutations are associated with normal stature and early onset osteoarthritis. Only a few non-glycine missense mutations have been reported and among these, the arginine to cysteine substitutions predominate and these mutations cause some unusual disorders which may be described as Stickler-like but have short stature and brachydactyly. Stickler syndrome type I (STL1) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL2A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type I.
Stickler syndrome type II (STL2) occurs due to mutations of the COL11A1 gene on chromosome 1p21 16, 17, 18. Patients with another condition called Marshall syndrome can have mutations of COL11A1 gene also, but patients with Stickler syndrome type II have a milder physical feature with less prominent facial dysmorphism than patients with Marshall syndrome. Patients with Stickler syndrome type II have less pronounced midfacial flattening and the nasal bridge better developed than seen in patients with Marshall syndrome. Myopia and retinal degeneration are not always present. Cataracts and more severe early onset hearing loss are more common in Stickler type II than in patients with Stickler type I. Stickler syndrome type II (STL2) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type II. There is also a autosomal recessive variety of Stickler syndrome type II (STL2) which has been identified in 3 people with very severe deafness.
Stickler syndrome type III (STL3) has been described as the non-eye form of Stickler syndrome, affecting the joints and hearing without involving the eyes. Stickler syndrome type III (STL3) is caused by mutations of the COL11A2 gene on chromosome 6p21.3 16, 17, 18. Stickler syndrome type III (STL3) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A2 gene that is inherited from one parent is enough to cause Stickler syndrome type III (STL3). Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type III (STL3). However, Stickler syndrome type III (STL3) is now considered the same disorder as heterozygous oto-spondylo-megaepiphyseal dysplasia (OSMED).
A mutation in a fourth gene, COL9A1, located on chromosome 6q13, has been identified in three reported intermarried families in Turkey and Morocco with Stickler syndrome type IV (STL4). The inheritance pattern is autosomal recessive meaning for a child to inherit Stickler syndrome type IV (STL4), the child need to receive two copies of a mutated COL9A1 gene, one from each parent. If a person only inherits one copy of the mutated COL9A1 gene, they are a carrier and usually do not show symptoms of Stickler syndrome type IV (STL4).
Stickler syndrome type V (STL5) is thought to be caused by COL9A2, located on chromosome 1p33. This has been described in one intermarried family in India. The inheritance pattern is autosomal recessive.
Mutations of COL9A3 have recently been reported in three brothers in an intermarried Moroccan family with features of Stickler syndrome and intellectual disability.
Stickler syndrome has also been subdivided based on the vitreous phenotype resulting from mutations in the various loci. However, it has been reported that it is difficult for most ophthalmologists to classify the type of vitreous anomalies in the patients with Stickler syndrome.
Stickler syndrome signs and symptoms
Eyes
- Short-sight or nearsightedness (myopia)
- Abnormal appearance of the vitreous gel.
- High risk of retinal detachments (tearing of the lining of the eye), which may affect both eyes.
- Cataracts
- Glaucoma
One of the first signs in Stickler syndrome is nearsightedness (myopia), in which objects close by are seen clearly but objects that are far away appear blurry. Myopia may vary from mild to severe in Stickler syndrome, but generally is not progressive and does not get worse 19. Myopia may be detectable shortly after birth, but the onset varies and may not develop until adolescence or even adulthood in some cases 19.
Bones and Joints
- Hyper-mobile (over flexible) joints and/or stiff joints.
- Early joint disease leading to osteoarthritis and joint replacements at a younger age
Facial Features
- A full cleft, submucous or high arched palate and/or bifid uvula
- Micrognathia – where the lower jaw is shorter than the other resulting in poor contact between the chewing surfaces of the upper and lower teeth. These symptoms are similar to those found in Pierre Robin sequence. It is reported that 30-40% of children diagnosed with Pierre Robin sequence are later re-diagnosed as having Stickler syndrome.
- Other facial characteristics include a flat face with a small nose and little or no nasal bridge. Appearance tends to improve with age
Hearing
- Hearing loss ( sensorineural and or conductive. The degree varies in affected individuals and may become more severe over time.
- Glue ear in childhood caused by cleft palate.
Other symptoms
These may include curvature of the spine (scoliosis), and because of sight and hearing problems, some learning difficulties may be experienced. Many people within the support group, especially children, complain of chronic fatigue.
A condition similar to Stickler syndrome, called Marshall syndrome, is characterized by a distinctive facial appearance, eye abnormalities, hearing loss, and early-onset arthritis. Marshall syndrome can also include short stature. Some researchers have classified Marshall syndrome as a variant of Stickler syndrome, while others consider it to be a separate disorder.
No studies to determine the prevalence of Stickler syndrome have been undertaken 2. However, an approximate incidence of Stickler syndrome among newborns can be estimated from data regarding the incidence of Robin sequence in newborns (1 per 10,000 to 1 per 14,000 newborns) and the percentage of these newborns who subsequently develop signs or symptoms of Stickler syndrome (35%). These data suggest that the incidence of Stickler syndrome among neonates is approximately 1 per 7,500 to 1 per 9,000 newborns 20. Stickler syndrome affects males as well as females.
Stickler is believed to be the most common connective tissue syndrome in the United States and Europe, but one of the rarest to be diagnosed. Most sufferers have such minor symptoms that they do not seek a diagnoses. Those who become patients are generally not correctly diagnosed. One study found a 53% error in original diagnosis of patients found in retrospect to have Stickler. A lot of patients are only diagnosed with one symptom and called, for example, arthritic or near-sighted.
The treatment of Stickler syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists including: geneticist, pediatrician and/or internist, orthopedic surgeon, rheumatologist, ophthalmologist and retina specialist, otolaryngologist, audiologist, plastic surgeon, orthodontist and other healthcare professionals may need to systematically and comprehensively plan an affected patient’s treatment.
Early identification of Stickler syndrome is important because it allows for surveillance and prompt treatment of associated abnormalities such as retinal detachment or skeletal malformations. Patients with ocular forms of Stickler syndrome are restricted from contact sports due to the risk of retinal detachment 1. Retinal detachment requires prompt surgery to preserve vision. Retinal detachment can recur even after successful surgery. Some physicians recommend prophylactic cryotherapy in certain cases to reduce the risk of developing retinal detachment.
Affected individuals should be made aware of the symptoms of retinal detachment so they can immediately have their eyes evaluated (ophthalmologic assessment) and treated if necessary. Surgery may also be necessary to remove cataracts.
Corrective lenses (glasses or contact lenses) are used to treat myopia.
Individuals with Stickler syndrome and Pierre-Robin sequence may require a tracheostomy (a procedure in which a tube is placed through a surgical opening in the neck) to prevent breathing (respiratory) difficulties. Surgery may also be required to fix various craniofacial abnormalities (e.g., cleft palate. micrognathia) that can contribute to breathing difficulties.
Orthodonture may be necessary to correct dental malalignment.
Patients with sensorineural or mixed hearing loss may require hearing aids. Hearing aids may be of benefit for certain individuals. A myringotomy, a surgical procedure in which a small incision is made in the eardrum and small tubes are inserted, may be used to treat glue ear. Various anti-inflammatory medications and sometimes prescription pain medications may be used to treat joint disease in individuals with Stickler syndrome. In mild cases, short-term relief may be provided from cortisone injections. Surgical correction of joint abnormalities may be necessary including joint replacement surgery such as a total hip or knee replacement. Surgery may also be necessary for skeletal malformations including abnormal curvature of the spine.
Physical therapy may prove beneficial in some cases.
Special education and other services may be helpful for children with learning disabilities due to hearing or vision problems.
Genetic counseling may be of benefit for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Stickler syndrome
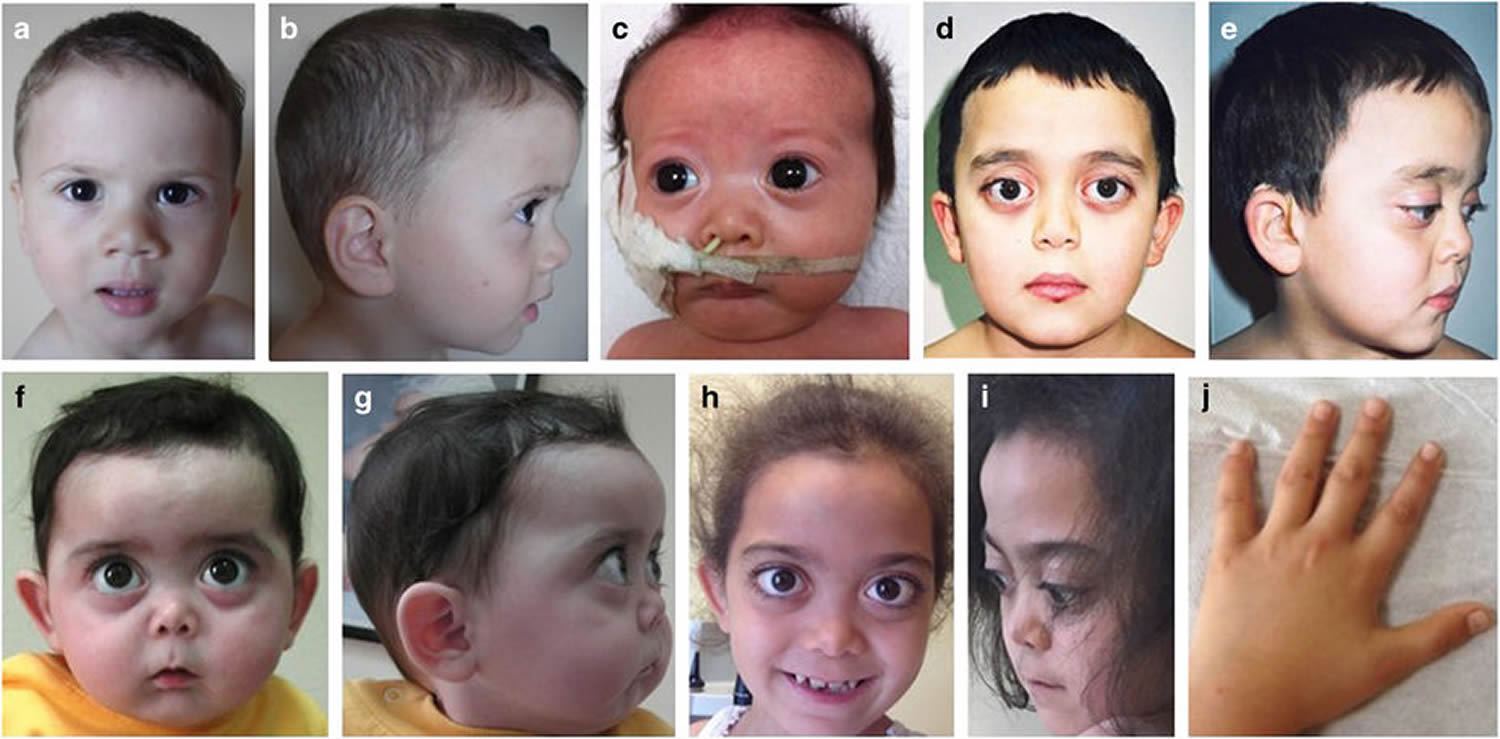
Note: Physical appearance of the patients. (a, b) Case 1, age 1.5 years. Mild depression of the nasal bridge and micrognathia. (c) Case 2, age 1 month. Buphthalmic eyes, hypertelorism, bilateral epicanthus, flat face, depressed nasal bridge, short stubby nose, and micro-retrognathia. (d, e) Case 4, age 8.5 years. Proptotic eyes, flat face with mild frontal bossing, depressed nasal bridge, and short nose. (f, g) Case 3, age 9 months. Buphthalmic eyes, flat face with frontal bossing, midfacial hypoplasia, depressed nasal bridge, short nose with anteverted nares, long philtrum, and micro-retrognathia. (h–j) Case 3, age 9 years. (h, i) High-frontal area, big proptotic eyes, long palpebral fissures, depressed nasal bridge, short nose, long philtrum, irregular teeth order, micrognathia, and dry rough hairs. (j) Small hands with brachydactyly.
Figure 2. Stickler syndrome type 1
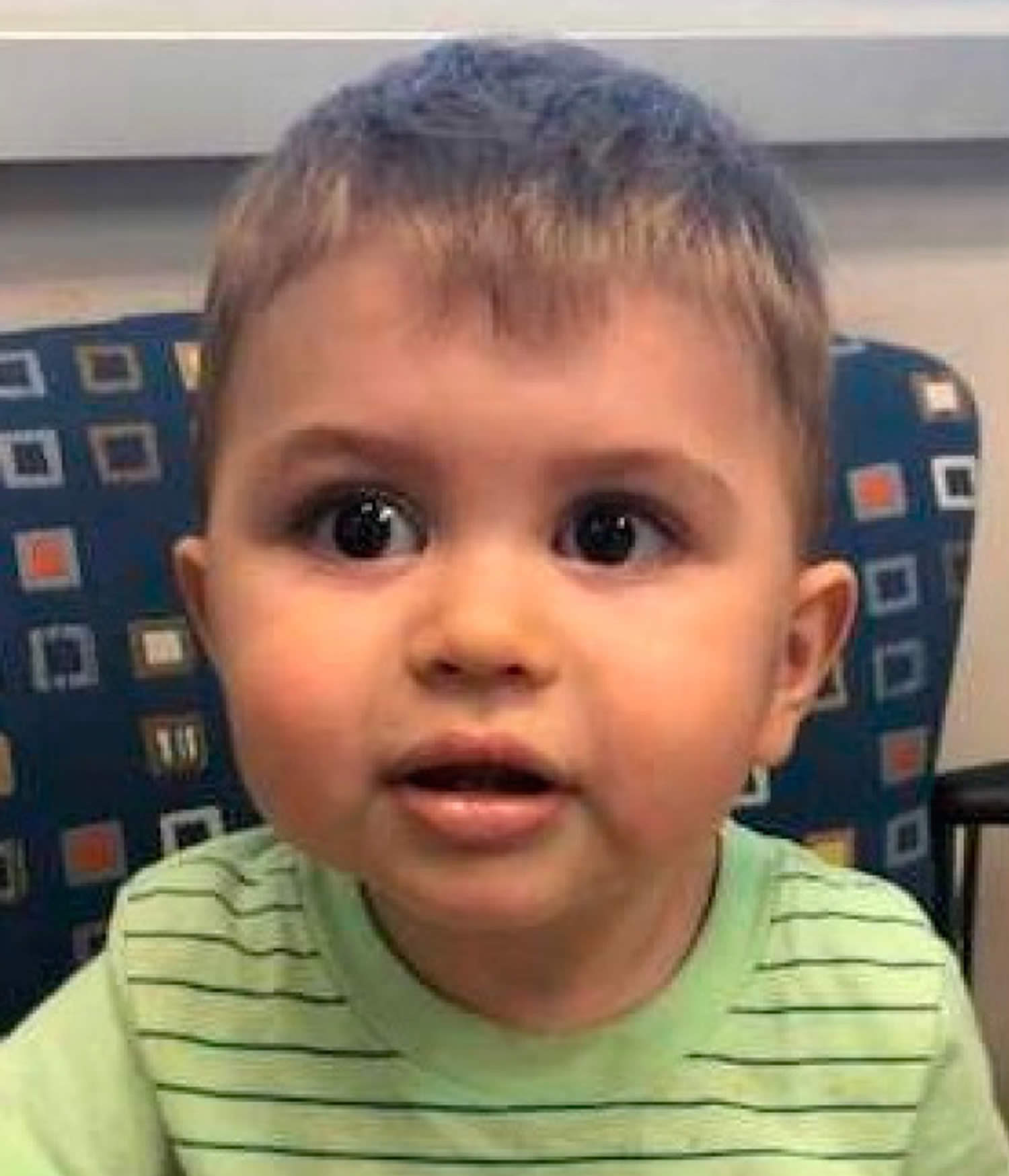
Footnotes: A child with classic Stickler syndrome type I (STL1 ) due to mutations in COL2A1 gene showing the characteristic midface hypoplasia and micrognathia are present.
[Source 13 ]Figure 3. Stickler syndrome joint hypermobility
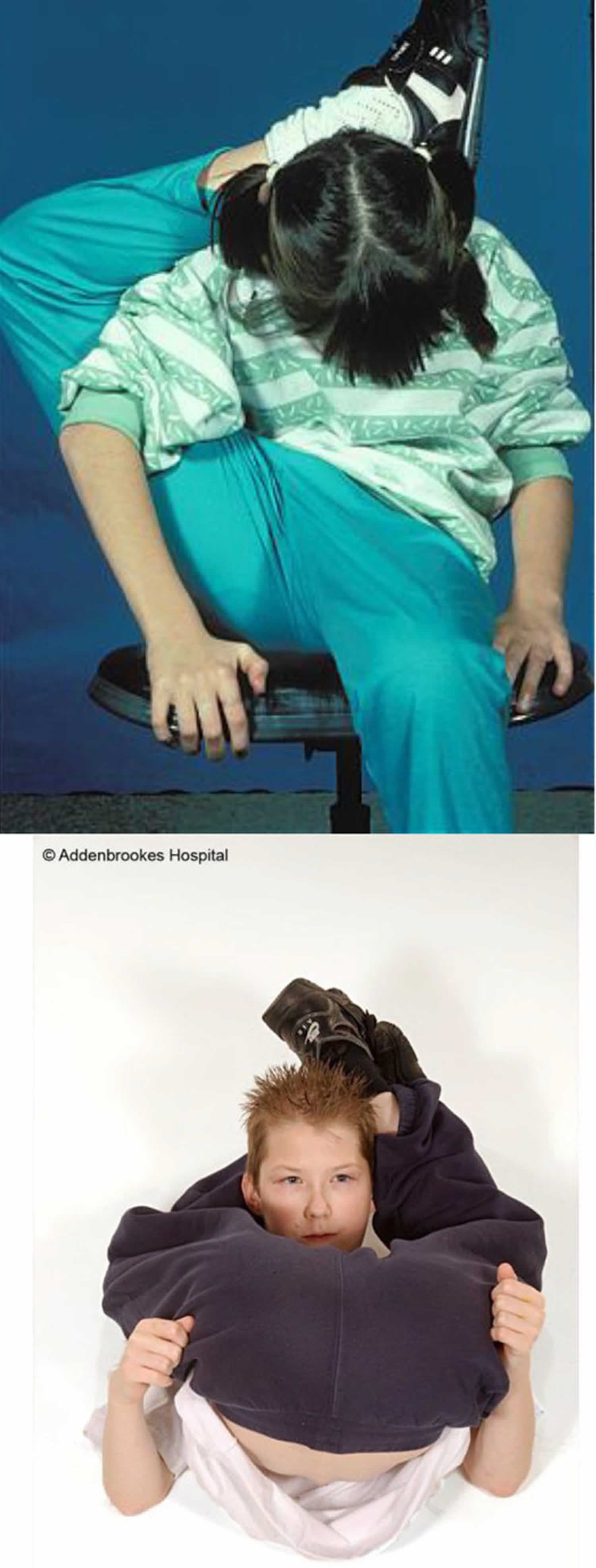
Figure 4. Stickler syndrome eye features
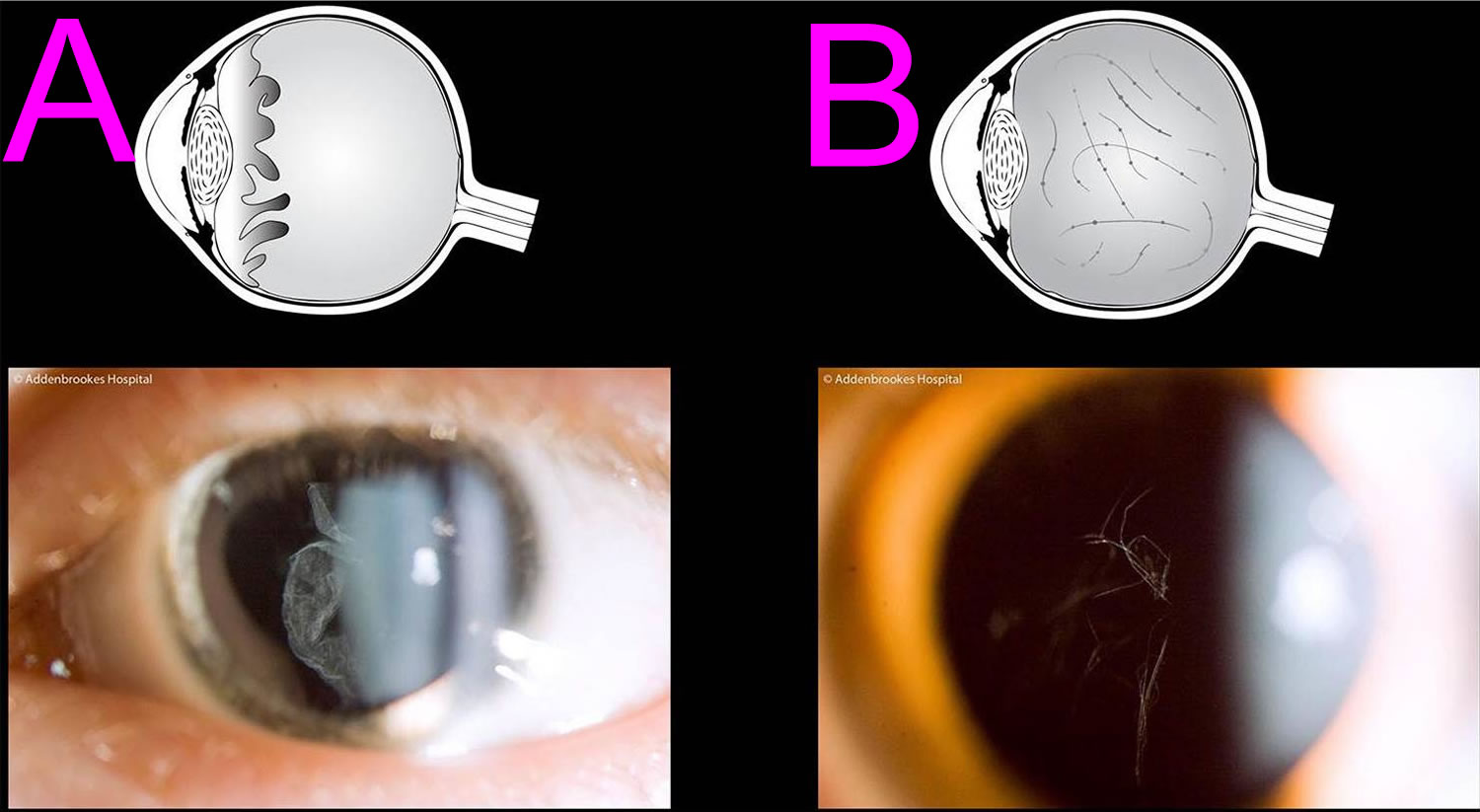
Footnotes: The pathognomonic hallmark of all but one of the sub-groups of Stickler syndrome (the only exception being Stickler syndrome type 3 where there is no eye involvement) is a congenital abnormality of vitreous embryogenesis. The secondary vitreous is normally fully matured by 10–14 weeks of intra-uterine growth and the embryogenic abnormalities in Stickler syndrome provide the clinician with a crucial sign on which to make the diagnosis. Stickler syndrome vitreoretinal abnormalities vary with the type of Stickler syndrome, sometimes described as membranous, beaded, or hypoplastic. Stickler syndrome Type 1 characteristically displays an optically empty-appearing central vitreous cavity with a membranous vitreous component within the vestigial gel in the retrolental space. Stickler syndrome Type 2 typically displays a beaded vitreous with irregular, thickened lamellae. Stickler syndrome Type 2 may less commonly have a hypoplastic vitreous which appears optically empty or with irregular lamellae architecture 22. (A) Membranous congenital vitreous anomaly (Stickler syndrome Type 1 haploinsufficiency mutations COL2A1 gene); (B) Beaded congenital vitreous anomaly (Stickler syndrome Type 2 COL11A1 dominant negative mutations).
[Source 21 ]Figure 5. Stickler syndrome cataract
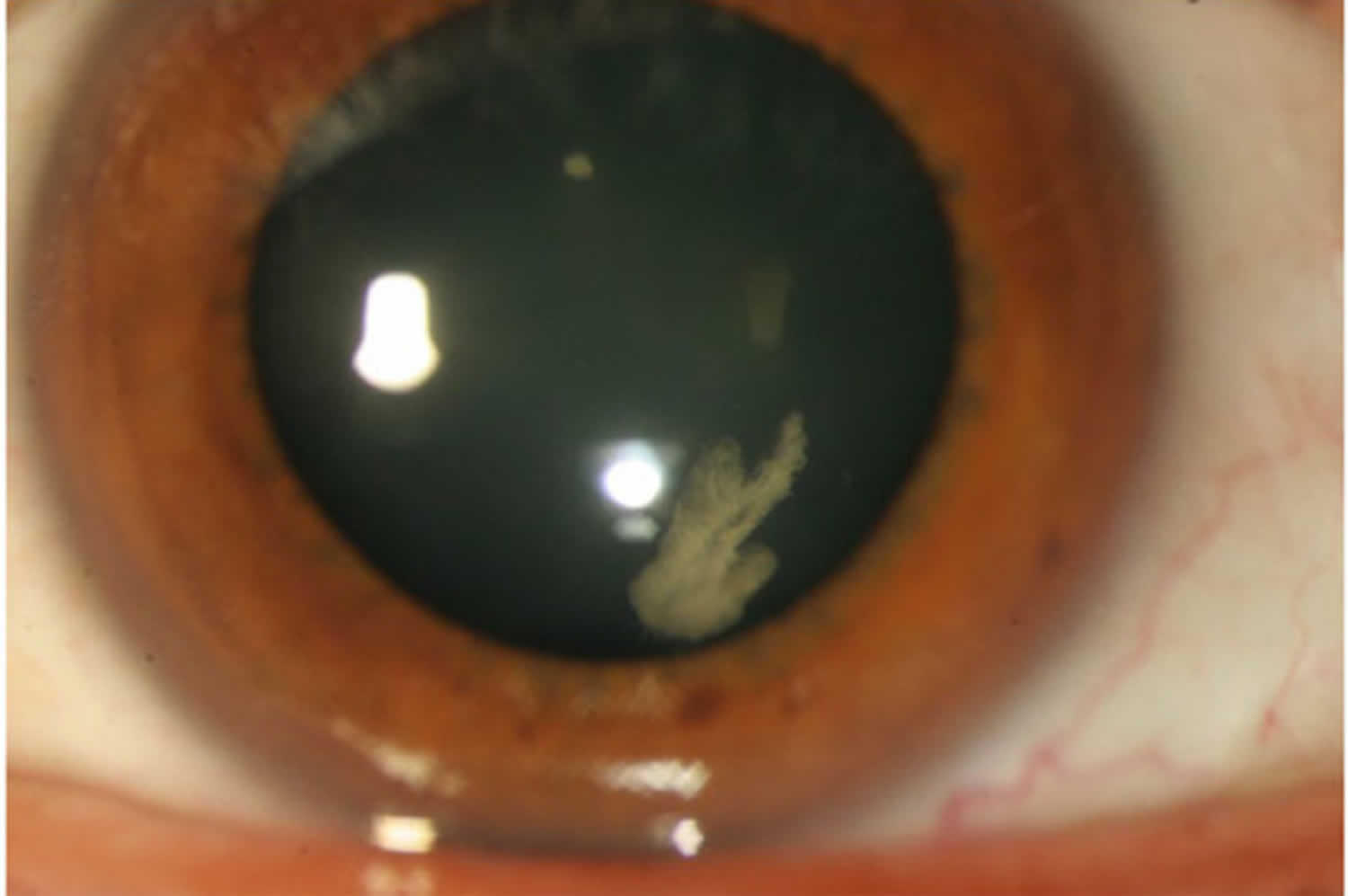
Footnotes: Congenital quadrantic lamellar cataract is well recognised and can be a useful diagnostic marker although it can be present in both Stickler syndrome type 1 and type 2 and does not therefore distinguish between sub-groups in the way that the differing vitreous features allow.
[Source 21 ]Figure 6. Stickler syndrome type 1 retinal radial lattice degeneration
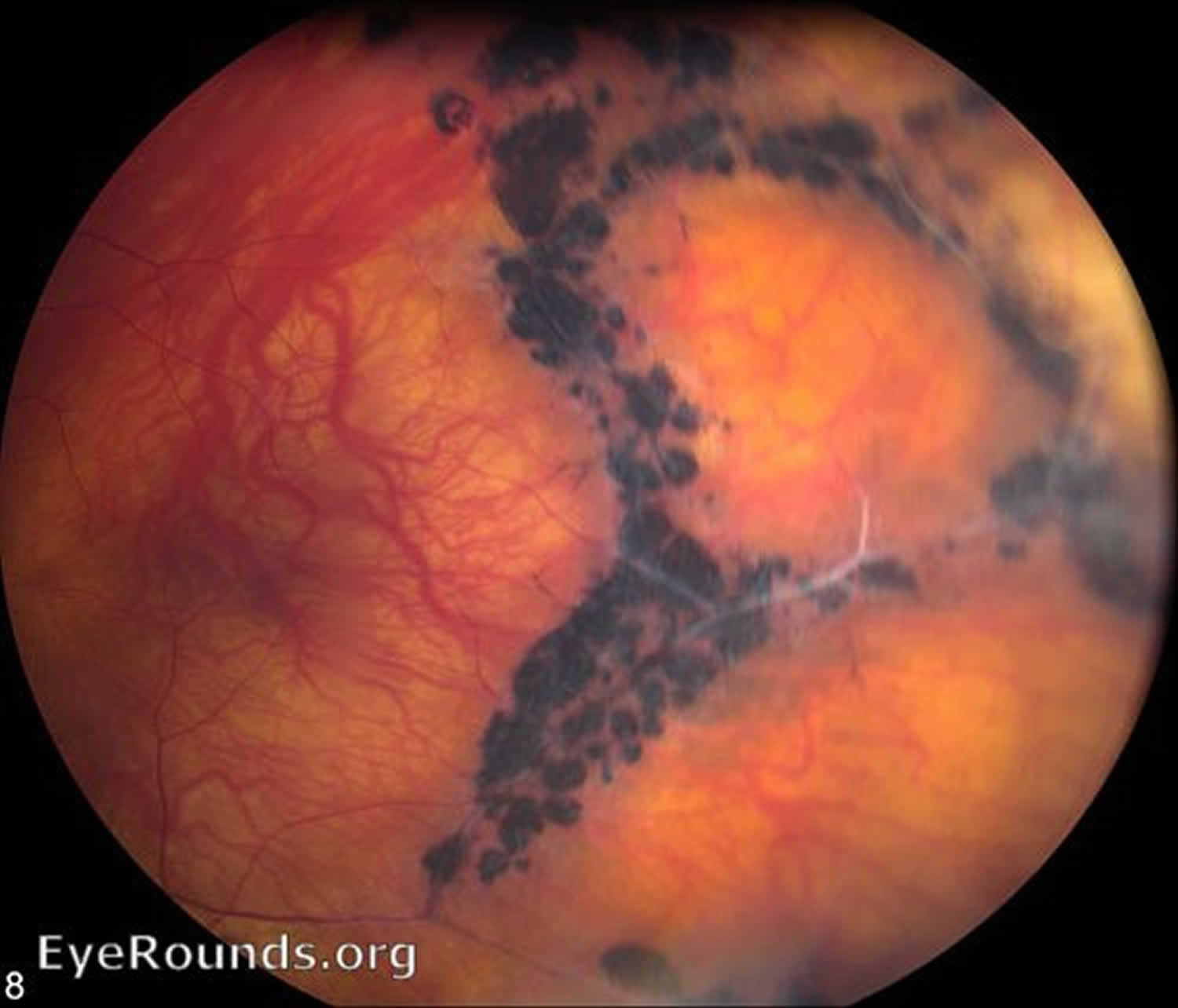
Footnotes: Extensive retinal radial lattice degeneration in Stickler syndrome Type 1 with COL2A1 gene mutation. The patient has had a retinal detachment in this eye with repair via scleral buckle and cryotherapy, seen on the temporal aspect in this photo.
[Source 23 ]Stickler syndrome types
Table 2. Stickler syndrome types
| Stickler syndrome | Gene | Cytogenetic location | Distinguishing features |
|---|---|---|---|
| Type 1 | COL2A1 | 12q13.11 | Type 1. Membranous congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, deafness, arthropathy, cleft palate High risk of blindness |
| Ocular only | COL2A1 | 12q13.11 | Type 1. Membranous congenital vitreous anomaly, retinal detachment congenital megalophthalmos. No systemic features. High risk of blindness |
| Type 2 | COL11A1 | 1p21.1 | Beaded type 2 congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, deafness, arthropathy, cleft palate |
| Type 2 Recessive | COL11A1 | 1p21.1 | Autosomal recessive, Beaded congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, cleft palate, profound severe congenital deafness |
| Type 3 | COL11A2 | 6p21.32 | Non-ocular Stickler |
| Type 4 | COL9A1 | 6q13 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 5 | COL9A2 | 1p34.2 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 6 | COL9A3 | 20q13.33 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 7 | BMP4 | Hypoplastic vitreous, retinal detachment deafness, arthropathy, palate abnormality, renal dysplasia | |
| Type 8 | LOXL3 | 2p13.1 | Recessive inheritance Congenital myopia, hypoplastic vitreous, palate abnormality, Arthropathy Normal facies Normal hearing |
Stickler syndrome type 1
Stickler syndrome type I (STL1) is responsible for approximately 80% to 90% of reported Stickler syndrome cases (most common form of Stickler syndrome) and presents with a wide variety of symptoms affecting the eye, ear, facial appearance, palate and musculoskeletal system 15, 12. Most will have ‘full’ Stickler syndrome affecting the sight, joints, hearing and any mid-line clefting. Findings show those with this anomaly have an increased incidence of cleft abnormalities. Stickler syndrome type I (STL1) is caused to mutations over the entire COL2A1 gene on chromosome 12q13.11 16, 17, 18. These mutations cause loss of function of the COL2A1 gene. The majority of these mutations are associated with normal stature and early onset osteoarthritis. Only a few non-glycine missense mutations have been reported and among these, the arginine to cysteine substitutions predominate and these mutations cause some unusual disorders which may be described as Stickler-like but have short stature and brachydactyly. Stickler syndrome type I (STL1) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL2A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type I.
Stickler syndrome type 2
Stickler syndrome type II (STL2) occurs due to mutations of the COL11A1 gene on chromosome 1p21 16, 17, 18. Patients with another condition called Marshall syndrome can have mutations of COL11A1 gene also, but patients with Stickler syndrome type II have a milder physical feature with less prominent facial dysmorphism than patients with Marshall syndrome. Patients with Stickler syndrome type II have less pronounced midfacial flattening and the nasal bridge better developed than seen in patients with Marshall syndrome. Myopia and retinal degeneration are not always present. Cataracts and more severe early onset hearing loss are more common in Stickler type II than in patients with Stickler type I. Stickler syndrome type II (STL2) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type II. There is also a autosomal recessive variety of Stickler syndrome type II (STL2) which has been identified in 3 people with very severe deafness.
Stickler syndrome type 3
Stickler syndrome type III (STL3) has been described as the non-eye form of Stickler syndrome, affecting the joints and hearing without involving the eyes. Stickler syndrome type III (STL3) is caused by mutations of the COL11A2 gene on chromosome 6p21.3 16, 17, 18. Stickler syndrome type III (STL3) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A2 gene that is inherited from one parent is enough to cause Stickler syndrome type III (STL3). Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type III (STL3). However, Stickler syndrome type III (STL3) is now considered the same disorder as heterozygous oto-spondylo-megaepiphyseal dysplasia (OSMED).
Stickler syndrome type 4
A mutation in a fourth gene, COL9A1, located on chromosome 6q13, has been identified in three reported intermarried families in Turkey and Morocco with Stickler syndrome type 4 or STL4.The inheritance pattern is autosomal recessive.
Stickler syndrome type 5
Stickler syndrome type 5 (STL5) is thought to be caused by COL9A2, located on chromosome 1p33. This has been described in one intermarried family in India. The inheritance pattern is autosomal recessive.
Stickler syndrome type 6
Mutations of COL9A3 have recently been reported in three brothers in an intermarried Moroccan family with features of Stickler syndrome and intellectual disability.
Stickler syndrome has also been subdivided based on the vitreous phenotype resulting from mutations in the various loci. However, it has been reported that it is difficult for most ophthalmologists to classify the type of vitreous anomalies in the patients with Stickler syndrome.
Stickler syndrome life expectancy
Because the symptoms of Stickler syndrome are variable, it can be difficult to predict what the long-term outlook is for people who have Stickler syndrome. There is an increased risk for eye problems associated with Stickler syndrome including retinal detachment and cataracts. These symptoms can lead to vision loss. People with Stickler syndrome may also experience arthritis before 40-years-old. In general, people with Stickler syndrome have typical intelligence and can function well in society. Some people do not know they have Stickler syndrome until another family member is diagnosed because the symptoms can be relatively mild 2.
Stickler syndrome causes
Mutations in several genes cause the different types of Stickler syndrome 16, 17, 18. Between 80 and 90 percent of all cases are classified as Stickler syndrome type I and are caused by mutations in the COL2A1 gene on the long (q) arm of chromosome 12 (12q13.11). Another 10 to 20 percent of cases are classified as Stickler syndrome type II and result from mutations in the COL11A1 gene on the short arm (p) of chromosome 1 (1p21). Marshall syndrome, which may be a variant of Stickler syndrome, is also caused by COL11A1 gene mutations. Stickler syndrome type III is caused by COL11A2 gene on the short arm (p) of chromosome 6 (6p21.3) and Stickler syndrome type IV results from mutations in COL9A1 gene located on the long (q) arm of chromosome 6 (6q13).
Stickler syndrome type V (STL5) is thought to be caused by COL9A2 gene, located on the short arm (p) of chromosome 1 (1p33). This has been described in one intermarried family in India. The inheritance pattern is autosomal recessive.
Mutations of COL9A3 have recently been reported in three brothers in an intermarried Moroccan family with features of Stickler syndrome and intellectual disability.
To date, more than 1,000 individuals have been identified with Stickler syndrome due to a mutations in one of the genes listed in Table 1.
All of the genes associated with Stickler syndrome provide instructions for making components of collagens, which are complex molecules that give structure and strength to the connective tissues that support the body’s joints and organs. Mutations in any of these genes impair the production, processing, or assembly of collagen molecules. Defective collagen molecules or reduced amounts of collagen impair the development of connective tissues in many different parts of the body, leading to the varied features of Stickler syndrome.
Not all individuals with Stickler syndrome have mutations in one of the known genes. Researchers believe that mutations in other genes may also cause this condition, but those genes have not been identified.
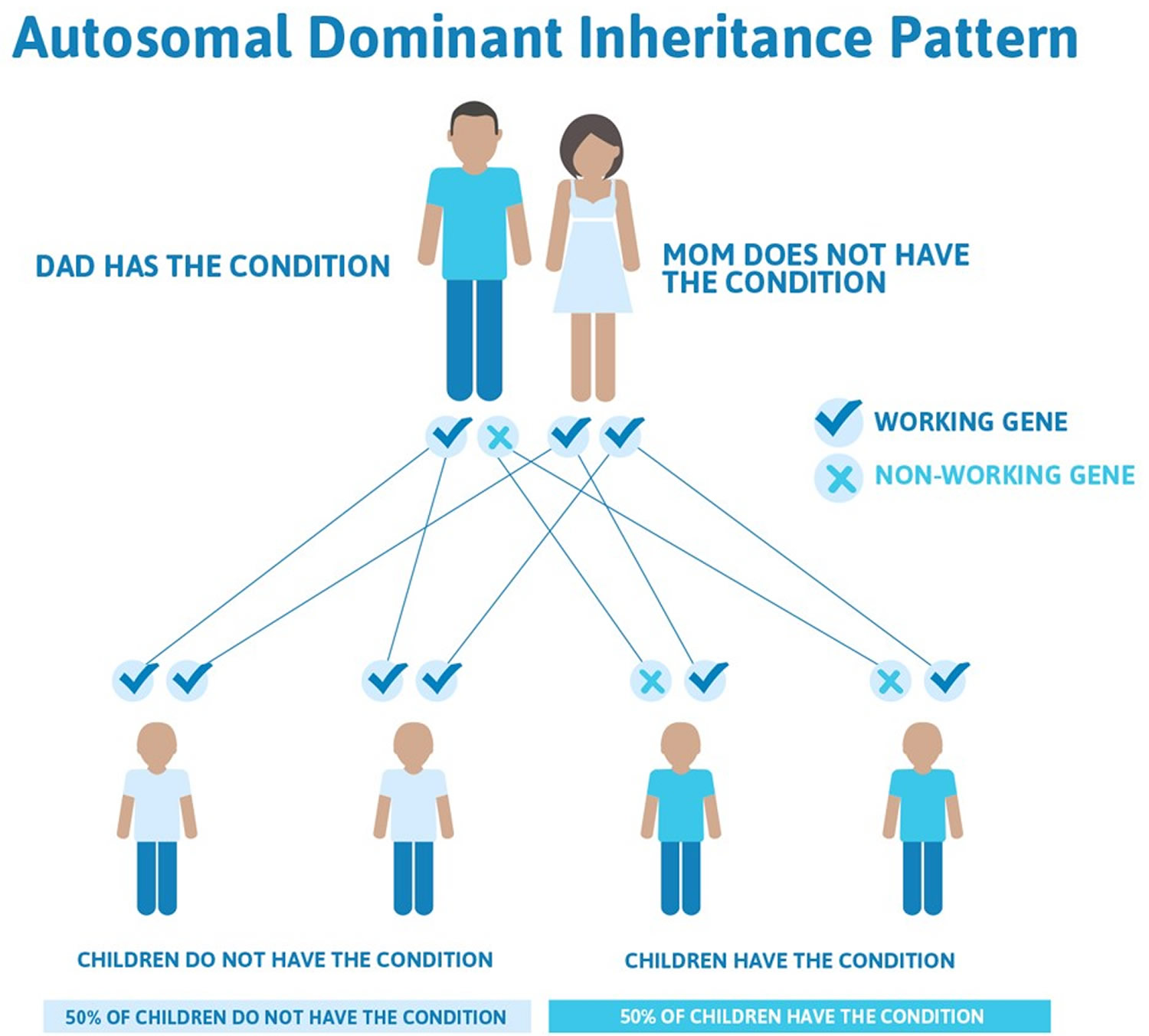
Stickler syndrome types I, II, and III are inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. In some cases, an affected person inherits a gene mutation from one affected parent. Other cases result from new mutations. These cases occur in people with no history of Stickler syndrome in their family. Marshall syndrome also typically has an autosomal dominant pattern of inheritance.
This means that only one copy of one of the genes causing Stickler syndrome types I, II, and III has a pathogenic variant. You inherit one copy of every gene from your mother and the other from your father. When a person who has Stickler syndrome types I, II, and III has children, for each child there is a:
- 50% chance that the child will inherit the gene with a pathogenic variant, meaning he or she will have Stickler syndrome
- 50% chance that the child will inherit the working copy of the gene, meaning he or she will not have Stickler syndrome
In some cases, people who have an autosomal dominant form of Stickler syndrome are the first people to be diagnosed in the family. This may be because they inherited the genetic change from a parent, but the parent has mild symptoms of the syndrome and was never diagnosed. Most people who have an autosomal dominant form of Stickler syndrome inherited the genetic change from a parent 2. In other cases, the genetic change may be new in the person who was diagnosed with Stickler syndrome. Genetic changes that are new in a person are called de novo 2.
Figure 7. Stickler syndrome types I, II, and III are inherited in an autosomal dominant inheritance pattern
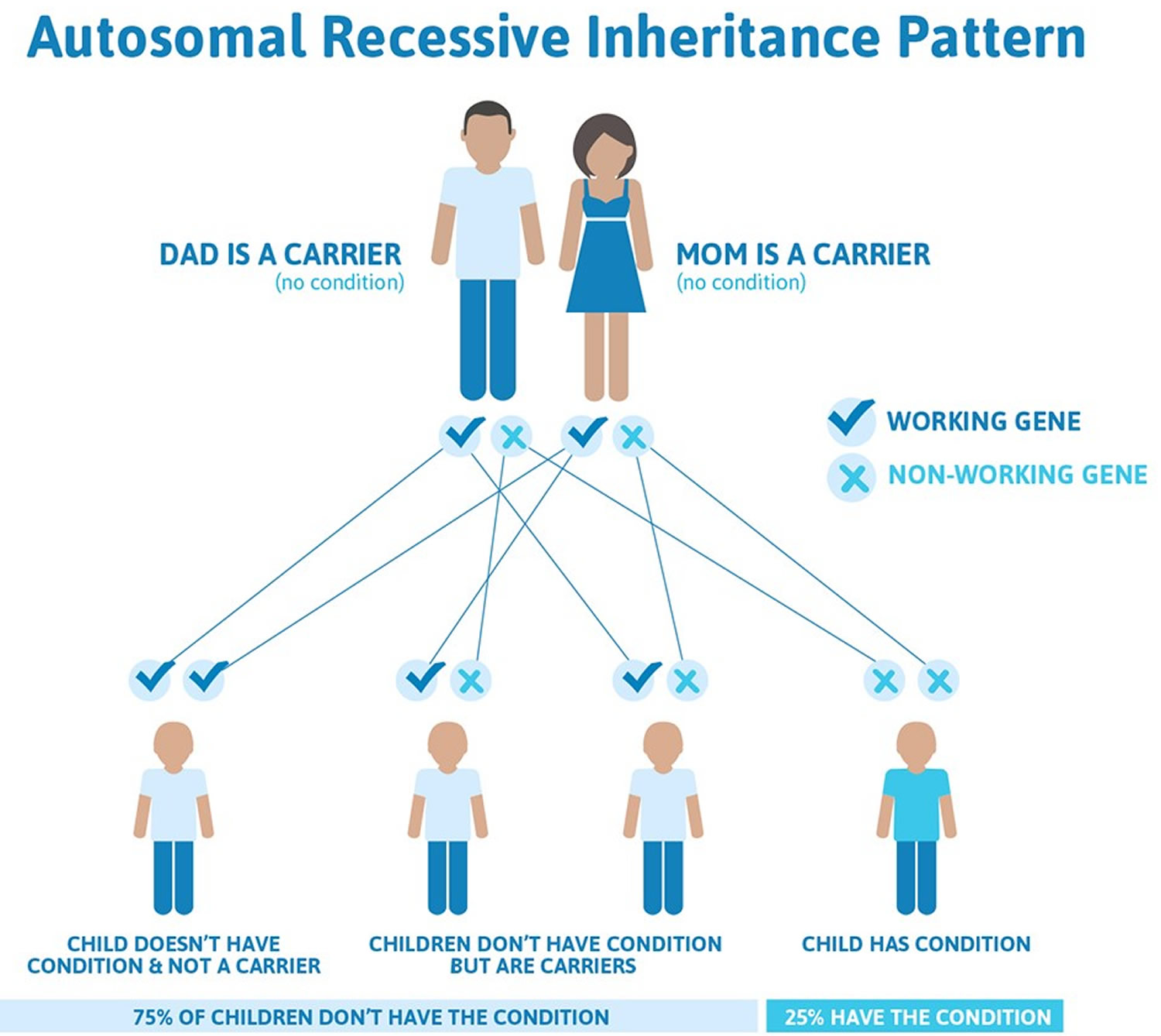
Stickler syndrome types IV, V, and VI are inherited in an autosomal recessive pattern. Autosomal recessive inheritance means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
This means that both copies of one of these genes must have a pathogenic variant for a person to have signs of Stickler syndrome types IV, V, and VI. People who only have one changed copy of a gene that causes an autosomal recessive form of Stickler syndrome are known as carriers. Carriers do not have signs or symptoms of the syndrome. When two carriers of Stickler syndrome have children together, for each child there is a:
- 25% chance that the child will inherit both changed copies of the gene, so he or she has Stickler syndrome
- 50% chance that the child will inherit only one changed copy of the gene, so he or she is a carrier of the syndrome like each of the parents
- 25% chance that the child will inherit both working copies of the gene, so he or she does not have Stickler syndrome and is not a carrier of the syndrome
Stickler syndrome shows a characteristic known as variable expressivity. This means that people with Stickler syndrome types IV, V, and VI can have different signs and symptoms of the syndrome, even among members of the same family. However, anyone with a pathogenic variant that causes Stickler syndrome is expected to have some symptoms of the syndrome. This is called full penetrance 2.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 8. Stickler syndrome types IV, V, and VI are inherited in an autosomal recessive inheritance pattern
Stickler syndrome symptoms
Stickler syndrome is a multisystem connective tissue disorder that can affect the craniofacies, eyes, inner ear, skeleton, and joints.
Stickler syndrome type I (STL1) is responsible for approximately 70% of reported Stickler syndrome cases (most common form of Stickler syndrome) and presents with a wide variety of symptoms affecting the eye, ear, facial appearance, palate and musculoskeletal system. Most will have ‘full’ Stickler syndrome affecting the sight, joints, hearing and any mid-line clefting. Findings show those with this anomaly have an increased incidence of cleft abnormalities. Stickler syndrome type I (STL1) is caused to mutations over the entire COL2A1 gene on chromosome 12q13.11 16, 17, 18. These mutations cause loss of function of the COL2A1 gene. The majority of these mutations are associated with normal stature and early onset osteoarthritis. Only a few non-glycine missense mutations have been reported and among these, the arginine to cysteine substitutions predominate and these mutations cause some unusual disorders which may be described as Stickler-like but have short stature and brachydactyly. Stickler syndrome type I (STL1) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL2A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type I.
Stickler syndrome type II (STL2) occurs due to mutations of the COL11A1 gene on chromosome 1p21 16, 17, 18. Patients with another condition called Marshall syndrome can have mutations of COL11A1 gene also, but patients with Stickler syndrome type II have a milder physical feature with less prominent facial dysmorphism than patients with Marshall syndrome. Patients with Stickler syndrome type II have less pronounced midfacial flattening and the nasal bridge better developed than seen in patients with Marshall syndrome. Myopia and retinal degeneration are not always present. Cataracts and more severe early onset hearing loss are more common in Stickler type II than in patients with Stickler type I. Stickler syndrome type II (STL2) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type II. There is also a autosomal recessive variety of Stickler syndrome type II (STL2) which has been identified in 3 people with very severe deafness.
Stickler syndrome type III (STL3) has been described as the non-eye form of Stickler syndrome, affecting the joints and hearing without involving the eyes. Stickler syndrome type III (STL3) is caused by mutations of the COL11A2 gene on chromosome 6p21.3 16, 17, 18. Stickler syndrome type III (STL3) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A2 gene that is inherited from one parent is enough to cause Stickler syndrome type III (STL3). Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type III (STL3). However, Stickler syndrome type III (STL3) is now considered the same disorder as heterozygous oto-spondylo-megaepiphyseal dysplasia (OSMED).
A mutation in a fourth gene, COL9A1, located on chromosome 6q13, has been identified in three reported intermarried families in Turkey and Morocco with Stickler syndrome type IV (STL4). The inheritance pattern is autosomal recessive meaning for a child to inherit Stickler syndrome type IV (STL4), the child need to receive two copies of a mutated COL9A1 gene, one from each parent. If a person only inherits one copy of the mutated COL9A1 gene, they are a carrier and usually do not show symptoms of Stickler syndrome type IV (STL4).
Stickler syndrome type V (STL5) is thought to be caused by COL9A2, located on chromosome 1p33. This has been described in one intermarried family in India. The inheritance pattern is autosomal recessive.
Mutations of COL9A3 have recently been reported in three brothers in an intermarried Moroccan family with features of Stickler syndrome and intellectual disability.
Stickler syndrome has also been subdivided based on the vitreous phenotype resulting from mutations in the various loci. However, it has been reported that it is difficult for most ophthalmologists to classify the type of vitreous anomalies in the patients with Stickler syndrome.
Craniofacial findings
Craniofacial findings include a flat facial profile or an appearance that is often referred to as a “scooped out” face. This profile is caused by underdevelopment of the maxilla and nasal bridge, which can cause telecanthus and epicanthal folds. Midface retrusion is most pronounced in infants and young children; older individuals may have a normal facial profile. Often the nasal tip is small and upturned, making the philtrum appear long.
Micrognathia is common and may be associated with cleft palate as part of the Pierre Robin sequence (micrognathia, cleft palate, glossoptosis). The degree of micrognathia may compromise the upper airway, necessitating tracheostomy.
Cleft palate may be seen in the absence of micrognathia.
Stickler syndrome eye features
Stickler syndrome eye findings include high myopia (>−3 diopters) that is non-progressive and detectable in the newborn period and may also develop degeneration of the thick jelly-like fluid (vitreous) that fills the center of the eyes and the thin layer of nerve cells (retina) that lines the back of the eye (vitreoretinal degeneration) 24. The retina senses light and converts it into nerve signals, which are then relayed to brain through the optic nerve. Vitreoretinal degeneration may cause tiny specks (floaters) that seem to float around obstructing a person’s field of vision. Vitreoretinal degeneration also places individuals with Stickler syndrome at risk for retinal detachment, which can affect one or both eyes.
Two types of vitreous abnormalities are observed:
- Type 1 (“membranous”), which is much more common, is characterized by a persistence of vestigial vitreous gel in the retrolental space that is bordered by a folded membrane.
- Type 2 (“beaded”), much less common, is characterized by sparse and irregularly thickened bundles throughout the vitreous cavity.
The Stickler syndrome eye findings run true within families 24.
Posterior chorioretinal atrophy was described by Vu et al 25 in a family with vitreoretinal dystrophy, a novel pathogenic variant in COL2A1, and systemic features of Stickler syndrome, suggesting that individuals with Stickler syndrome may have posterior pole chorioretinal changes in addition to the vitreous abnormalities.
Retinal detachment occurs when the retina pulls away or is separated (detaches) from the underlying tissue. In some cases, small tears may occur in the retina as well. Symptoms of retinal detachment include an increase in the number of floaters in the eye, increased blurriness of vision, sudden flashes of light and a sudden decrease in vision as if a curtain or veil is pulled over a portion of a person’s field of vision. Retinal detachment can cause significant loss of vision or blindness if left untreated. Retinal detachment can occur at any age.
Additional eye abnormalities associated with Stickler syndrome include clouding (opacity) of the lenses of the eyes (cataracts), crossed eyes (strabismus), and abnormal curvature to the cornea (the clear portion of the eye through which light passes) or lens of the eye (astigmatism), which can contribute to blurred vision. A small percentage of individuals with Stickler syndrome, approximately 5-10 percent, may develop glaucoma, a condition in which increased pressure within the eye causes characteristic damage to the optic nerve, which relays signals from the retina to the brain.
Hearing loss
Hearing loss is common in Stickler syndrome. The degree of hearing loss is variable and may be progressive. The degree of hearing loss may vary greatly from one individual to another and can range from mild to significant.
Some degree of sensorineural hearing impairment (hearing loss due to impaired ability of the auditory nerves to transmit sensory input to the brain) that is typically high-tone and often subtle is found in 40% of individuals 24. The exact mechanism is unclear, although it is related to the expression of type II and IX collagen in the inner ear 26. Overall sensorineural hearing loss in Stickler syndrome type I is typically mild and not significantly progressive; it is less severe than that reported for Stickler syndrome types II and III.
Conductive hearing loss or hearing loss due to failure of sound waves being conducted through the middle ear can also be seen in Stickler syndrome. This may be secondary to recurrent middle ear infections (otitis media) that are often associated with cleft palate and/or may be secondary to a defect of the ossicles of the middle ear. Some individuals may develop the accumulation of thick, sticky fluid behind the eardrum (glue ear). People with Stickler syndrome can have hypermobility of the middle ear bones.
Hearing loss is common in Stickler syndrome can also be mixed hearing loss from both sensorineural hearing and conductive hearing loss.
Facial features
Individuals with Stickler syndrome often have distinctive facial features including mid-facial hypoplasia with abnormally flat cheek bones and nasal bridge, small nose, long upper lip (philtrum), prominent eyes, and small chin.
Affected individuals may also have Pierre-Robin sequence, an assortment of abnormalities that may occur as a distinct syndrome or as part of another underlying disorder. Pierre-Robin sequence is characterized by an unusually small jaw (micrognathia), downward displacement or retraction of the tongue (glossoptosis), and incomplete closure of the roof of the mouth (cleft palate, sub-mucous cleft palate or bifid uvula). Often babies with Pierre-Robin sequence and glossoptosis obstruct their airway when placed on their backs, and it may be recommended that they sleep prone due to this risk. Patients with very small jaws might be recommended to have surgery to extend their jaw forward.
Cleft palate may also occur as an isolated finding. The various craniofacial features may give the face a flattened appearance, but these features usually become less distinctive as affected children grow older. Certain facial features such as cleft palate can cause feeding or breathing difficulties in some children.
Dental anomalies such as failure of the upper and lower teeth to meet when biting down (malocclusion) may also occur.
Skeletal features
Skeletal malformations are a common finding in individuals with Stickler syndrome. Skeletal manifestations are early-onset arthritis, short stature relative to unaffected siblings, and radiographic findings consistent with mild spondyloepiphyseal dysplasia. Some individuals have a slender body habitus, but without tall stature.
Abnormally flexible or lax (hypermobile) joints (double jointedness) that may make them prone to joint dislocation, is sometimes seen in young individuals, becomes less prominent or resolves completely with age 24. Joint pain and stiffness upon rest are frequent findings, and many individuals develop inflammation of the joints during the third or fourth decade of life (early-onset osteoarthritis).
Early-onset arthritis is common and may be severe, leading to the need for surgical joint replacement even as early as the third or fourth decade. More commonly, the arthropathy is mild, and affected individuals often do not complain of joint pain unless specifically asked. However, nonspecific complaints of joint stiffness can be elicited even from young children.
Spinal abnormalities are also common observed in Stickler syndrome that result in chronic back pain are abnormal sideways curvature of the spine (scoliosis), front-to-back curvature of the spine (kyphosis), forward displacement of one vertebra over another, usually the 4th lumbar vertebra over the 5th or the 5th over the sacrum (spondylolisthesis), endplate abnormalities and platyspondylia 27. Spinal abnormalities associated with Stickler may become progressively worse and may be associated with back pain.
Chest deformities such as pectus excavatum (depression of the chest bone) and carinatum (prominent chest bone) can occur in individuals with Stickler syndrome.
Additional findings may occur in some cases including diminished muscle tone (hypotonia), abnormally long, slender fingers (arachnodactyly), flat feet (pes planus), and osteochondritis deformans of the hips (Legg-Calve-Perthes disease), a degenerative hip disorder with childhood onset.
Other features of Stickler syndrome
Intelligence is unaffected in children with Stickler syndrome, but some children may develop learning disabilities because of hearing and vision abnormalities.
Some studies have seemed to indicate that the prevalence of mitral valve prolapse (MVP) is greater in individuals with Stickler syndrome (4%) than in the general population (2%). However, other studies seem to show that this is not the case. The mitral valve is located between the left upper and left lower chambers (left atrium and left ventricle) of the heart. Mitral valve prolapse occurs when one or both of the flaps (cusps) of the mitral valve bulge or collapse backward (prolapse) into the left upper chamber (atrium) of the heart. In some cases, this may allow leakage or the backward flow of blood from the left lower chamber of the heart (ventricle) back into the left atrium (mitral valve regurgitation). In some cases, no associated symptoms are apparent (asymptomatic). However, in other cases, mitral valve prolapse can result in chest pain, abnormal heart rhythms (arrhythmias), fatigue, and dizziness.
Mitral valve prolapse has been reported in nearly 50% of individuals with Stickler syndrome in one series 28; diagnosis of Stickler syndrome was made on clinical features prior to the identification of the involved genes. A later study 29 reported mitral valve prolapse on echocardiogram in only one of 25 individuals with Stickler syndrome and a COL2A1 pathogenic variant. Ahmad et al 30 screened a group of 75 individuals with molecularly confirmed Stickler syndrome and found no individuals with clinical or echocardiographic evidence of significant mitral valve or other valve abnormality. It was suggested that among those with Stickler syndrome, the prevalence of mitral valve prolapse may be similar to that in the general population. No additional studies reviewing cardiac findings in Stickler syndrome have been reported.
Stickler syndrome diagnosis
Stickler syndrome can sometimes be diagnosed based on your child’s medical history and a physical exam, additional tests are needed to determine the severity of the symptoms and help direct treatment decisions.
Stickler syndrome should be suspected in individuals with a combination of the following findings:
- Cleft palate (open cleft, submucous cleft, or bifid uvula)
- Characteristic facial features including malar hypoplasia, broad or flat nasal bridge, and micro/retrognathia
- Vitreous changes or retinal abnormalities (lattice degeneration, retinal hole, retinal detachment or retinal tear)
- High-frequency sensorineural hearing loss
- Skeletal findings including:
- Slipped epiphysis or Legg-Perthes-like disease
- Scoliosis, spondylolisthesis, or Scheuermann-like kyphotic deformity
- Osteoarthritis before age 40
- An independently affected first-degree relative
Establishing the Diagnosis
The diagnosis of Stickler syndrome is established in a index case who meets the proposed clinical diagnostic criteria and/or has a heterozygous pathogenic variant in COL2A1, COL11A1, or COL11A2 or biallelic pathogenic variants in COL9A1, COL9A2, or COL9A3.
Clinical Diagnostic Criteria
Clinical diagnostic criteria have been proposed for type 1 Stickler syndrome (in which individuals have the membranous type of vitreous abnormality; see Clinical Description) but not validated [Rose et al 2005]. The proposed criteria are based on assigning points for clinical features, family history data, and molecular data.
Stickler syndrome should be considered in individuals with ≥5 points and absence of features suggestive of an alternative diagnosis. At least one finding should be a major (2-point) manifestation (denoted by *).
Abnormalities (2-pt maximum per category)
- Orofacial
- Cleft palate* (open cleft, submucous cleft, or bifid uvula): 2 points
- Characteristic facial features (malar hypoplasia, broad or flat nasal bridge, and micro/retrognathia): 1 point
- Ocular. Characteristic vitreous changes or retinal abnormalities* (lattice degeneration, retinal hole, retinal detachment or retinal tear): 2 points
- Auditory
- High-frequency sensorineural hearing loss*: 2 points
- Age <20 years: threshold ≥20 dB at 4-8 Hz
- Age 20-40 years: threshold ≥30 dB at 4-8 Hz
- Age >40 years: threshold ≥40 dB at 4-8 Hz
- Hypermobile tympanic membranes: 1 point
- Skeletal
- Femoral head failure (slipped epiphysis or Legg-Perthes-like disease): 1 point
- Radiographically demonstrated osteoarthritis before age 40: 1 point
- Scoliosis, spondylolisthesis, or Scheuermann-like kyphotic deformity: 1 point
Family history/molecular data
Independently affected first-degree relative in a pattern consistent with autosomal dominant inheritance or presence of a COL2A1, COL11A1, or COL11A2 pathogenic variant associated with Stickler syndrome**: 1 point
* Denotes major manifestation
** Does not account for families with autosomal recessive Stickler syndrome
Molecular genetic testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing:
- Serial single-gene testing can be considered based on the individual’s clinical findings and family history; however, findings should not be used to exclude specific testing:
- COL2A1 may be tested first in individuals with ocular findings including type 1 “membranous” congenital vitreous anomaly and milder hearing loss.
- COL11A1 may be tested first in individuals with typical ocular findings including type 2 “beaded” congenital vitreous anomaly and significant hearing loss.
- COL11A2 may be tested for in individuals with craniofacial and joint manifestations and hearing loss but without ocular findings.
- COL9A1, COL9A2, and COL9A3 may be tested for in individuals with possible autosomal recessive inheritance.
Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- A multigene panel that includes COL2A1, COL11A1, COL11A2, COL9A1, COL9A2, COL9A3 and other genes of interest may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene varies by laboratory and over time. (2) Some multigene panels may include genes not associated with the condition discussed; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Stickler syndrome treatment
There’s no cure for Stickler syndrome. Treatment addresses the signs and symptoms of the disorder that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists including: geneticist, pediatrician and/or internist, orthopedic surgeon, rheumatologist, ophthalmologist and retina specialist, otolaryngologist, audiologist, plastic surgeon, orthodontist and other healthcare professionals may need to systematically and comprehensively plan an affected patient’s treatment.
To establish the extent of disease and needs in an individual diagnosed with Stickler syndrome, the following evaluations are recommended:
- Evaluation of palate by a craniofacial specialist
- Baseline ophthalmologic examination
- Baseline audiogram
- Directed history to elicit complaints suggestive of mitral valve prolapse, such as episodic tachycardia and chest pain. If symptoms are present, referral to a cardiologist should be made.
- Consultation with a clinical geneticist and/or genetic counselor.
Affected individuals should be advised to avoid activities that may lead to traumatic retinal detachment (e.g., contact sports) 1.
Some physicians recommend avoiding physical activities that involve high impact to the joints to delay the onset of the arthropathy. While this recommendation seems logical, there are no data to support it 1.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Craniofacial
Infants with Robin sequence need immediate attention from specialists in otolaryngology and pediatric critical care, as they may require tracheostomy to ensure a competent airway. It is recommended that evaluation and management occur in a comprehensive craniofacial clinic that provides all the necessary services, including otolaryngology, plastic surgery, oral and maxillofacial surgery, pediatric dentistry, orthodontics, and medical genetics.
In most individuals, micrognathia tends to become less prominent over time, allowing for removal of the tracheostomy. However, in some individuals, significant micrognathia persists, causing orthodontic problems. In these individuals, a mandibular advancement procedure is often required to correct the malocclusion.
Stickler syndrome eye
Refractive errors should be corrected with spectacles.
Individuals with Stickler syndrome should be advised of the symptoms associated with retinal detachment and the need for immediate evaluation and treatment when such symptoms occur.
Retinal detachment is a common cause of visual loss in patients with Stickler syndrome, occurring in up to 65% of affected patients 31. Stickler syndrome is the most common inherited cause of rhegmatogenous retinal detachment 32. Retinal detachment causing visual loss most typically occurs in young adulthood, but may occur within the first five years of life. Patients are particularly at risk of developing a giant retinal tear, which typically occurs during posterior vitreous detachment. Associated retinal detachments are notoriously difficult to manage and patients are prone to development of proliferative vitreoretinopathy 33. Treatment with primary vitrectomy is generally more successful than scleral buckling procedures and has been suggested to the procedure of choice in Stickler syndrome-related retinal detachment. However, a recent study comparing pars plana vitrectomy (PPV), scleral buckle (SB), and combination pars plana vitrectomy and scleral buckle found initial scleral buckle had better visual acuity and anatomic outcomes in treating retinal detachments in syndromes with optically empty vitreous 34.
Prophylactic treatment of peripheral retina using 360° cryotherapy or circumferential laser is effective in reducing rates of retinal detachment 31. Both treatment modalities have been retrospectively compared to observation and both are apparently effective in significantly decreasing risk of retinal detachment. However, there has not been a study directly comparing the two modalities, and meta-analyses have not shown one to be superior to the other 35, 36. The Cambridge Prophylactic Cryotherapy Protocol study in patients with Stickler syndrome type 1 found that untreated control patients had a 7.4-fold increased risk of retinal detachment compared to patients who had bilateral 360° transconjunctival prophylactic cryotherapy 37. The rate of retinal detachment was 53.6% in the bilateral control group and 8.3% in the bilateral prophylaxis group 37. A study of 360° extended vitreous base laser prophylaxis in Stickler syndrome found a significant reduction in the rate of retinal detachment with laser treatment (3% incidence of retinal detachment) as compared to no treatment (73% incidence of retinal detachment) 38. Additionally, this study found that eyes that received extended vitreous base laser had approximately 8 lines better vision on average compared to untreated eyes 38. In this study, laser was applied from the ora serrata to the equator for 360° with laser burn spacing between one half to 1 spot size 38. Morris et al. 39 reported successful prophylaxis of retinal detachment in 5 treated eyes of patients with Stickler Syndrome Type 2 using encircling grid laser from 2mm onto the pars plana to the ora and 4mm posteriorly, followed by extension of the laser posteriorly to and between the vortex vein ampullae 3 months later, to protect against more posterior retinal defects. It has been recommended that the option for prophylactic treatment be discussed with patients and their families in early life 33.
Patients with associated open-angle glaucoma may require surgical intervention 31. The availability of published data to guide management in such cases is limited; however, glaucoma in Stickler syndrome is presumed to be related to anterior segment dysgenesis and therefore angle surgery (goniotomy or trabeculotomy) may be beneficial. Filtering procedures remain viable options for refractory cases 31.
The typical cataract found in Stickler syndrome is of a congenital quadratic lamellar type 40. Such cataracts may or may not negatively impact vision; if decreased vision is present, removal may be performed with standard lensectomy or phacoemulsification. Patients are at an increased risk of both vitreous loss and post-operative retinal detachment; retinal breaks should be addressed prior to cataract surgery 41.
Audiologic
Otitis media may be a recurrent problem secondary to palatal abnormalities. Myringotomy tubes are often required.
The ultimate goal in the evaluation and treatment of a child with hereditary hearing loss and deafness is mainstream schooling. Research shows that diagnosis by age three months and habilitation by six months makes this goal possible for children with mild-moderate hearing loss.
Your child may need speech therapy if hearing loss interferes with his or her ability to learn how to pronounce certain sounds.
If your child has problems hearing, you may find that his or her quality of life is improved by wearing a hearing aid. Cochlear implantation in children with severe-to-profound deafness who are part of mainstream education leads to social functioning and educational attainment that is indistinguishable from normal-hearing peers 42.
Special education
Hearing or vision problems may cause learning difficulty in school, so special education services may be helpful.
Joints
Treatment of arthropathy is symptomatic and includes using over-the-counter anti-inflammatory medications before and after physical activity.
In some cases, physical therapy may help with mobility problems associated with joint pain and stiffness. Equipment such as braces, canes and arch supports also may help.
Surgery
- Tracheostomy. Newborns with very small jaws and displaced tongues may need a tracheostomy to create a hole in the throat so that they can breathe. The operation is reversed once the baby has grown large enough that his or her airway is no longer blocked.
- Jaw surgery. Surgeons can lengthen the lower jaw by breaking the jawbone and implanting a device that will gradually stretch the bone as it heals.
- Cleft palate repair. Babies born with a hole in the roof of the mouth (cleft palate) typically undergo surgery in which tissue from the roof of the mouth may be stretched to cover the cleft palate.
- Ear tubes. The surgical placement of a short plastic tube in the eardrum can help reduce the frequency and severity of ear infections, which are especially common in children who have Stickler syndrome.
- Eye surgeries. Surgeries to remove cataracts or procedures to reattach the lining of the back of the eye (retina) may be necessary to preserve vision.
- Joint replacement. Early-onset arthritis, particularly in the hips and knees, may necessitate joint replacement surgeries at a much younger age than is typical for the general population.
- Spinal bracing or fusion surgeries. Children who develop abnormal curves in their spines, such as those seen in scoliosis and kyphosis, may require corrective surgery. Milder curves often can be treated with a brace.
Home remedies
- Pain relievers. Over-the-counter drugs such as ibuprofen (Advil, Motrin IB) and naproxen sodium (Aleve) may help relieve joint swelling, stiffness and pain.
- Avoid contact sports. Strenuous physical activity may stress the joints, and contact sports, such as football, may increase the risk of retinal detachment.
- Seek educational help. Your child may have difficulty in school due to problems hearing or seeing. Your child’s teachers need to be aware of his or her special needs.
- Mortier G. Stickler Syndrome. 2000 Jun 9 [Updated 2023 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1302[↩][↩][↩][↩]
- Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. 2000 Jun 9 [Updated 2017 Mar 16]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1302/[↩][↩][↩][↩][↩][↩]
- Acke FRE, De Leenheer EMR. Hearing Loss in Stickler Syndrome: An Update. Genes (Basel). 2022 Sep 1;13(9):1571. doi: 10.3390/genes13091571[↩]
- Fernández-Pérez JJ, Mascaraque-Ruiz P, Martín Gómez C, Martínez-Caballero I, Otón T, Carmona L, Lara SL. Quality of Life in Children and Adolescents with Stickler Syndrome in Spain. Children (Basel). 2022 Aug 19;9(8):1255. doi: 10.3390/children9081255[↩]
- Stickler G.B., Hughes W., Houchin P. Clinical Features of Hereditary Progressive Arthro-Ophthalmopathy (Stickler Syndrome): A Survey. Genet. Med. 2001;3:192–196. doi: 10.1097/00125817-200105000-00008[↩]
- Stickler G., Belau P., Farrell F., Jones J., Pugh D., Steinberg A., Ward L. Hereditary Progessive Arthro-Ophthalmopathy. Mayo Clin. Proc. 1965;13:433–455. doi: 10.1017/s1120962300015900[↩][↩]
- Rose P.S., Levy H.P., Liberfarb R.M., Davis J., Szymko-Bennett Y., Rubin B.I., Tsilou E., Griffith A.J., Francomano C.A. Stickler Syndrome: Clinical Characteristics and Diagnostic Criteria. Am. J. Med. Genet. Part A. 2005;138:199–207. doi: 10.1002/ajmg.a.30955[↩][↩]
- Webb A.C., Markus A.F. The Diagnosis and Consequences of Stickler Syndrome. Br. J. Oral Maxillofac. Surg. 2002;40:49–51. doi: 10.1054/bjom.2001.0747[↩][↩]
- Alexander P, Fincham GS, Brown S, Collins D, McNinch AM, Poulson AV, Richards A, Martin H, Wareham N, Snead MP. Cambridge Prophylactic Protocol, Retinal Detachment, and Stickler Syndrome. N Engl J Med. 2023 Apr 6;388(14):1337-1339. doi: 10.1056/NEJMc2211320[↩]
- Karempelis P, Hagen M, Morrell N, Roby BB. Associated syndromes in patients with Pierre Robin Sequence. Int J Pediatr Otorhinolaryngol. 2020 Apr;131:109842. doi: 10.1016/j.ijporl.2019.109842[↩]
- Davies A, Davies A, Wren Y, Deacon S, Cobb A, McLean N, David D, Chummun S. Syndromes associated with Robin sequence: a national prospective cohort study. Arch Dis Child. 2023 Jan;108(1):42-46. doi: 10.1136/archdischild-2022-324722[↩]
- Hoornaert KP, Vereecke I, Dewinter C, et al. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010 Aug;18(8):872-80. doi: 10.1038/ejhg.2010.23. Erratum in: Eur J Hum Genet. 2010 Aug;18(8):881.[↩][↩][↩]
- Boothe M, Morris R, Robin N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. J Pers Med. 2020 Aug 27;10(3):105. doi: 10.3390/jpm10030105[↩][↩]
- Wang DD, Gao FJ, Hu FY, Zhang SH, Xu P, Wu JH. Mutation Spectrum of Stickler Syndrome Type I and Genotype-phenotype Analysis in East Asian Population: a systematic review. BMC Med Genet. 2020 Feb 10;21(1):27. doi: 10.1186/s12881-020-0963-z[↩][↩]
- Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J Med Genet. 1999 May;36(5):353-9. https://pmc.ncbi.nlm.nih.gov/articles/instance/1734362/pdf/v036p00353.pdf[↩][↩]
- Richards A.J., Baguley D.M., Yates J.R.W., Lane C., Nicol M., Harper P.S., Scott J.D., Snead M.P. Variation in the Vitreous Phenotype of Stickler Syndrome Can Be Caused by Different Amino Acid Substitutions in the X Position of the Type II Collagen Gly-X-Y Triple Helix. Am. J. Hum. Genet. 2000;67:1083–1094. doi: 10.1016/S0002-9297(07)62938-3[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Acke F.R., Swinnen F.K., Malfait F., Dhooge I.J., De Leenheer E.M.R. Auditory Phenotype in Stickler Syndrome: Results of Audiometric Analysis in 20 Patients. Eur. Arch. Otorhinolaryngol. 2016;273:3025–3034. doi: 10.1007/s00405-016-3896-6[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Snead M.P., McNinch A.M., Poulson A.V., Bearcroft P., Silverman B., Gomersall P., Parfect V., Richards A.J. Stickler Syndrome, Ocular-Only Variants and a Key Diagnostic Role for the Ophthalmologist. Eye. 2011;25:1389. doi: 10.1038/eye.2011.201[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Stickler Syndrome. https://rarediseases.org/rare-diseases/stickler-syndrome[↩][↩]
- Printzlau A, Andersen M. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J. 2004;41:47–52. https://www.ncbi.nlm.nih.gov/pubmed/14697070[↩]
- Snead M, Martin H, Bale P, Shenker N, Baguley D, Alexander P, McNinch A, Poulson A. Therapeutic and diagnostic advances in Stickler syndrome. Ther Adv Rare Dis. 2020 Dec 9;1:2633004020978661. doi: 10.1177/2633004020978661[↩][↩][↩][↩]
- Richards AJ, McNinch A, Martin H, Oakhill K, Rai H, Waller S, Treacy B, Whittaker J, Meredith S, Poulson A, Snead MP. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010 Jun;31(6):E1461-71. doi: 10.1002/humu.21257[↩]
- Radial Lattice Degeneration in Stickler’s syndrome. https://eyerounds.org/atlas/pages/Radial-lattice-Sticklers/index.htm#gsc.tab=0[↩]
- Snead M, Yates J. Clinical and molecular genetics of Stickler syndrome. Journal of Medical Genetics. 1999;36(5):353-359. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1734362/pdf/v036p00353.pdf[↩][↩][↩][↩]
- Vu CD, Brown J Jr, Korkko J, Ritter R 3rd, Edwards AO. Posterior chorioretinal atrophy and vitreous phenotype in a family with Stickler syndrome from a mutation in the COL2A1 gene. Ophthalmology. 2003;110:70–7. https://www.ncbi.nlm.nih.gov/pubmed/12511349[↩]
- Admiraal RJ, Brunner HG, Dijkstra TL, Huygen PL, Cremers CW. Hearing loss in the nonocular Stickler syndrome caused by a COL11A2 mutation. Laryngoscope. 2000;110:457–61. https://www.ncbi.nlm.nih.gov/pubmed/10718438[↩]
- Rose PS, Ahn NU, Levy HP, Ahn UM, Davis J, Liberfarb RM, Nallamshetty L, Sponseller PD, Francomano CA. Thoracolumbar spinal abnormalities in Stickler syndrome. Spine. 2001;26:403–9. https://www.ncbi.nlm.nih.gov/pubmed/11224888[↩]
- Liberfarb RM, Goldblatt A. Prevalence of mitral-valve prolapse in the Stickler syndrome. Am J Med Genet. 1986;24:387–92. https://www.ncbi.nlm.nih.gov/pubmed/3728560[↩]
- Liberfarb RM, Levy HP, Rose PS, Wilkin DJ, Davis J, Balog JZ, Griffith AJ, Szymko-Bennett YM, Johnston JJ, Francomano CA, Tsilou E, Rubin BI. The Stickler syndrome: genotype/phenotype correlation in 10 families with Stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genet Med. 2003;5:21–7. https://www.ncbi.nlm.nih.gov/pubmed/12544472[↩]
- Ahmad N, Richards AJ, Murfett HC, Shapiro L, Scott JD, Yates JR, Norton J, Snead MP. Prevalence of mitral valve prolapse in Stickler syndrome. Am J Med Genet. 2003;116A:234–7. https://www.ncbi.nlm.nih.gov/pubmed/12503098[↩]
- Stickler Syndrome. https://eyewiki.org/Stickler_Syndrome[↩][↩][↩][↩]
- Ang A, Poulson AV, Goodburn SF, Richards AJ, Scott JD, Snead MP. Retinal detachment and prophylaxis in type 1 Stickler syndrome. Ophthalmology. 2008 Jan;115(1):164-8. doi: 10.1016/j.ophtha.2007.03.059[↩]
- Retina and Vitreous, Section 12. Basic and Clinical Science Course, AAO, 2011-2012.[↩][↩]
- Taylor K, Su M, Richards Z, Mamawalla M, Rao P, Chang E. Outcomes in Retinal Detachment Repair and Laser Prophylaxis for Syndromes with Optically Empty Vitreous. Ophthalmol Retina. 2023 Jun 23:S2468-6530(23)00280-4. doi: 10.1016/j.oret.2023.06.012[↩]
- Carroll C, Papaioannou D, Rees A, Kaltenthaler E. The clinical effectiveness and safety of prophylactic retinal interventions to reduce the risk of retinal detachment and subsequent vision loss in adults and children with Stickler syndrome: a systematic review. Health Technol Assess. 2011 Apr;15(16):iii-xiv, 1-62. doi: 10.3310/hta15160[↩]
- Boysen KB, La Cour M, Kessel L. Ocular complications and prophylactic strategies in Stickler syndrome: a systematic literature review. Ophthalmic Genet. 2020 Jun;41(3):223-234. doi: 10.1080/13816810.2020.1747092[↩]
- Fincham GS, Pasea L, Carroll C, McNinch AM, Poulson AV, Richards AJ, Scott JD, Snead MP. Prevention of retinal detachment in Stickler syndrome: the Cambridge prophylactic cryotherapy protocol. Ophthalmology. 2014 Aug;121(8):1588-97. doi: 10.1016/j.ophtha.2014.02.022[↩][↩]
- Khanna S, Rodriguez SH, Blair MA, Wroblewski K, Shapiro MJ, Blair MP. Laser Prophylaxis in Patients with Stickler Syndrome. Ophthalmol Retina. 2022 Apr;6(4):263-267. doi: 10.1016/j.oret.2021.11.001[↩][↩][↩]
- Morris RE, Parma ES, Robin NH, Sapp MR, Oltmanns MH, West MR, Fletcher DC, Schuchard RA, Kuhn F. Stickler Syndrome (SS): Laser Prophylaxis for Retinal Detachment (Modified Ora Secunda Cerclage, OSC/SS). Clin Ophthalmol. 2021 Jan 6;15:19-29. doi: 10.2147/OPTH.S284441[↩]
- Snead MP, McNinch AM, Poulson AV, Bearcroft P, Silverman B, Gomersall P, Parfect V, Richards AJ. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (Lond). 2011 Nov;25(11):1389-400. doi: 10.1038/eye.2011.201[↩]
- Pediatric Ophthalmology and Strabismus, Section 6. Basic and Clinical Science Course, AAO, 2011-2012.[↩]
- Langereis M, Vermeulen A. School performance and wellbeing of children with CI in different communicative–educational environments. Int J Pediatr Otorhinolaryngol. 2015;79:834–9. https://www.ncbi.nlm.nih.gov/pubmed/25840945[↩]





{kind=link}