What is connective tissue disease
Connective tissue diseases is an umbrella term for a wide range of disorders that affect your body’s connective tissues, the fibers that are composed of collagen and elastin that support and anchor your organs and other structures in your body, such as tendons, ligaments, skin, cartilage, bones, fascia and blood vessels, which provide support and structure to other tissues and organs. Connective tissue diseases can be divided into 2 main types depending on its origin: (1) Inherited genetic disorders caused by gene defects like Marfan syndrome (a inherited genetic disorder that affects connective tissue most commonly the heart, eyes, blood vessels and skeleton; people with Marfan syndrome are usually tall and thin with unusually long arms, legs, fingers and toes) and Ehlers-Danlos syndrome (a group of 13 inherited genetic connective tissue disorders including joint hypermobility, skin hyperextensibility, and tissue fragility) and (2) Autoimmune disorders also called autoimmune diseases caused by the body’s immune system which normally helps protect your body from infection and disease begin attacking its own tissues (autoimmune) for unknown reasons such as Systemic Lupus Erythematosus (Lupus; an autoimmune disease that can affect many parts of the body, including the skin, joints, kidneys, and brain), Rheumatoid Arthritis (a chronic autoimmune inflammatory condition that primarily affects the joints, causing pain, swelling, and stiffness) and Scleroderma (an autoimmune disease that causes hardening and tightening of the skin and can also affect internal organs) 1. Other health experts also include sarcomas into this category of connective tissue disease. Sarcomas are cancerous (malignant) tumors that arise from cells that make up the connective tissues. Sarcomas are rare tumors that account for approximately 1% of tumors in humans. Soft tissue sarcomas can arise from fat, muscle, nerve, tendons and blood and lymph vessel tissue. For some sarcomas the tissue of origin is uncertain such as pleiomorphic undifferentiated sarcoma. Synovial sarcoma is a misnomer as it does not arise from synovial tissue (as was originally thought). Sarcomas can occur almost anywhere in the body, but the most common areas are the arms and legs, the back of the abdomen (retroperitoneum) and head and neck. Sarcomas can affect adults or children. These tumors are diverse with significantly different signs and symptoms, progression and often different treatment regimens. According to the World Health Organization (WHO) classification, there are more than 100 different histologic subtypes of soft tissue sarcomas. The exact, underlying cause of these tumors is not fully understood. Most likely, complex genetic and environmental factors play a role in their development.
Connective tissue diseases can affect many organs in your body including your heart, lungs, kidneys, joints and gastrointestinal tract, potentially leading to serious complications. Connective tissue disease symptoms can vary greatly but may include joint pain and stiffness, skin rashes, fatigue, shortness of breath, organ-specific problems and Raynaud’s phenomenon also known as Raynaud’s disease that causes decreased blood flow to your fingers and toes in response to cold temperatures or stress.
Connective tissue disease types
Connective tissue diseases can be divided into 3 types depending on its origin: (1) Inherited genetic disorders caused by gene defects like Marfan syndrome (a inherited genetic disorder that affects connective tissue most commonly the heart, eyes, blood vessels and skeleton; people with Marfan syndrome are usually tall and thin with unusually long arms, legs, fingers and toes) and Ehlers-Danlos syndrome (a group of 13 inherited genetic connective tissue disorders including joint hypermobility, skin hyperextensibility, and tissue fragility) and (2) Autoimmune disorders also called autoimmune diseases caused by the body’s immune system which normally helps protect your body from infection and disease attacking its own tissues (autoimmune) such as Systemic Lupus Erythematosus (Lupus; an autoimmune disease that can affect many parts of the body, including the skin, joints, kidneys, and brain), Rheumatoid Arthritis (a chronic autoimmune inflammatory condition that primarily affects the joints, causing pain, swelling, and stiffness) and Scleroderma (an autoimmune disease that causes hardening and tightening of the skin and can also affect internal organs). Sarcomas is another type of connective tissue disease. Sarcomas are cancerous (malignant) tumors that arise from cells that make up the connective tissues. Sarcomas are rare tumors that account for approximately 1% of tumors in humans. Soft tissue sarcomas can arise from fat, muscle, nerve, tendons and blood and lymph vessel tissue. For some sarcomas the tissue of origin is uncertain such as pleiomorphic undifferentiated sarcoma. Synovial sarcoma is a misnomer as it does not arise from synovial tissue (as was originally thought). Sarcomas can occur almost anywhere in the body, but the most common areas are the arms and legs, the back of the abdomen (retroperitoneum) and head and neck. Sarcomas can affect adults or children. These tumors are diverse with significantly different signs and symptoms, progression and often different treatment regimens. According to the World Health Organization (WHO) classification, there are more than 100 different histologic subtypes of soft tissue sarcomas. The exact, underlying cause of these tumors is not fully understood. Most likely, complex genetic and environmental factors play a role in their development.
Autoimmune connective tissue diseases
Autoimmune diseases are what many people think of when they think of connective tissue disease. In these conditions, your immune system generates chronic inflammation in some parts of your body. Chronic inflammation causes pain, swelling and, eventually, permanent damage to your tissues.
Some examples of autoimmune connective tissue disorders include:
Systemic Lupus Erythematosus (Lupus)
Figure 1. Systemic lupus erythematosus rash (systemic lupus erythematosus butterfly rash)
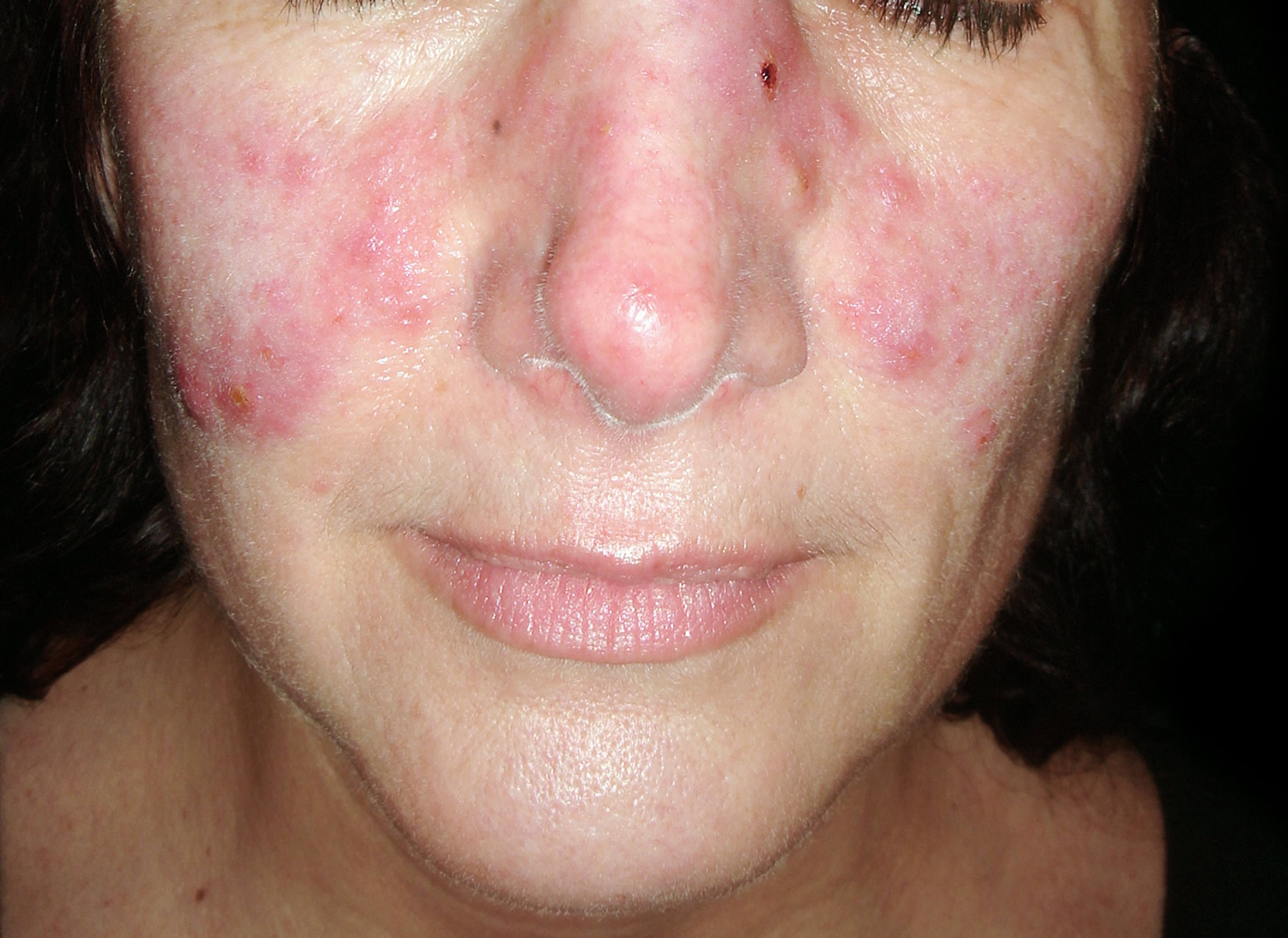
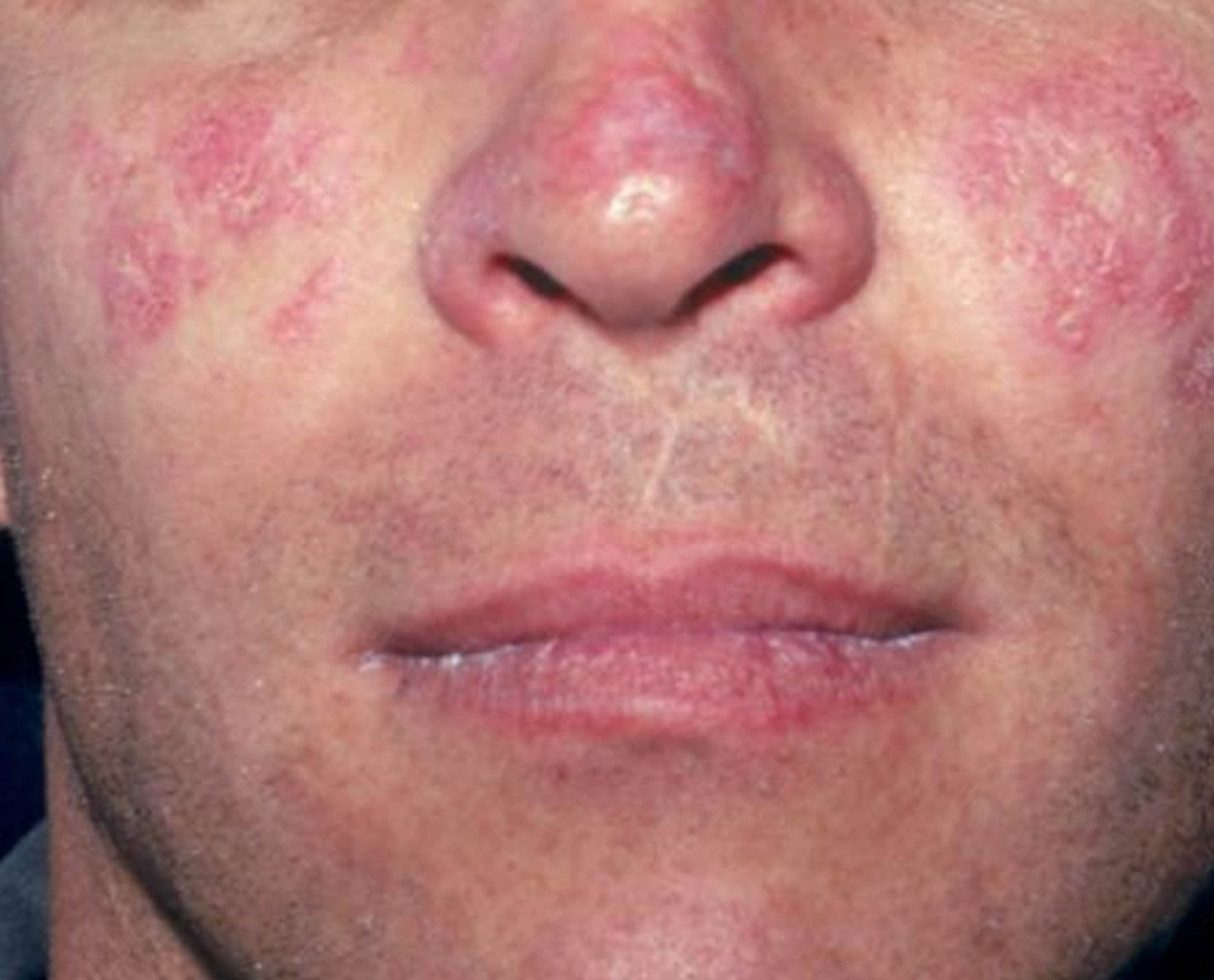
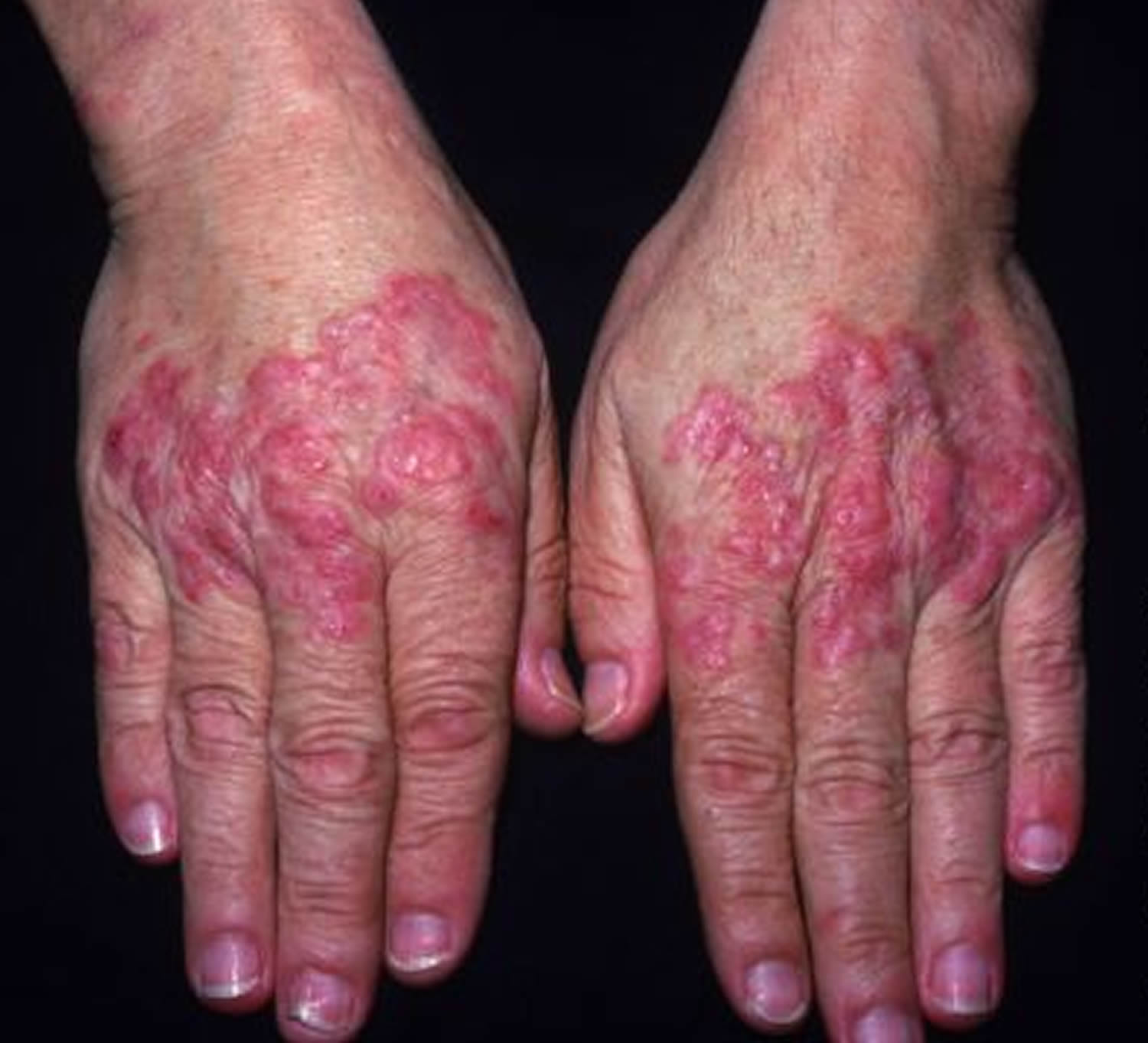
Figure 2. Systemic lupus erythematosus rash hand
No two cases of systemic lupus erythematosus (SLE) are exactly alike. Systemic lupus erythematosus (SLE) signs and symptoms may come on suddenly or develop slowly, may be mild or severe, and may be temporary or permanent. Most people with SLE have mild disease characterized by episodes called flares in which the signs and symptoms get worse (exacerbations) for a while, then improve or even disappear completely for a time (remissions). Overall, SLE gradually gets worse over time, and damage to the major organs of the body can be life-threatening.
The signs and symptoms of systemic lupus erythematosus (SLE) that you experience will depend on which body systems are affected by the disease. The most common signs and symptoms include:
- Fatigue (feeling tired all the time)
- Fever
- Joint pain, stiffness and swelling
- Joint pain, muscle pain or chest pain (especially when you’re taking a deep breath).
- Butterfly-shaped rash (butterfly rash) on the face that covers the cheeks and bridge of the nose or rashes elsewhere on the body
- Skin lesions that appear or worsen with sun exposure
- Fingers and toes that turn white or blue when exposed to cold or during stressful periods (Raynaud’s syndrome)
- Shortness of breath (dyspnea)
- Chest pain
- Dry eyes
- Headaches, confusion and memory loss
- Hair loss
- Mouth sores
- Swollen glands
- Blood clots
- Swelling in your arms, legs or on your face.
- Photosensitivity (sensitivity to sunlight).
- Dry eyes.
- Depression or other mental health conditions.
- Seizures.
- Anemia.
- Osteoporosis.
- Heart disease. Lupus can cause inflammation of your heart muscle (myocarditis), your arteries (arteritis) or heart membrane (pericarditis). The risk of cardiovascular disease and heart attacks increases greatly as well.
- Blood and blood vessels disease. Lupus may lead to blood problems, including a reduced number of healthy red blood cells (anemia) and an increased risk of bleeding or blood clotting. It can also cause inflammation of the blood vessels (vasculitis).
- Lungs. Having lupus increases your chances of developing an inflammation of the chest cavity lining (pleurisy), which can make breathing painful. Bleeding into lungs and pneumonia also are possible.
- Kidney disease. Lupus can cause serious kidney damage, and kidney failure is one of the leading causes of death among people with lupus.
- Brain and central nervous system complications. If your brain is affected by lupus, you may experience headaches, dizziness, behavior changes, vision problems, and even strokes or seizures. Many people with lupus experience memory problems and may have difficulty expressing their thoughts.
Having lupus also increases your risk of:
- Infection. People with lupus are more vulnerable to infection because both the disease and its treatments can weaken the immune system.
- Cancer. Having lupus appears to increase your risk of cancer; however, the risk is small.
- Bone tissue death (osteonecrosis). This occurs when the blood supply to a bone declines, often leading to tiny breaks in the bone and eventually to the bone’s collapse.
- Pregnancy complications. Women with lupus have an increased risk of miscarriage. Lupus increases the risk of high blood pressure during pregnancy (preeclampsia) and preterm birth. To reduce the risk of these complications, doctors often recommend delaying pregnancy until your disease has been under control for at least six months.
Systemic lupus erythematosus (SLE) may first appear as extreme tiredness (fatigue), a vague feeling of discomfort or illness (malaise), fever, loss of appetite, and weight loss. Most affected individuals also have joint pain, typically affecting the same joints on both sides of the body, and muscle pain and weakness.
About a third of people with SLE develop kidney disease (nephritis). Heart problems may also occur in SLE, including inflammation of the sac-like membrane around the heart (pericarditis) and abnormalities of the heart valves, which control blood flow in the heart. Heart disease caused by fatty buildup in the blood vessels (atherosclerosis), which is very common in the general population, is even more common in people with SLE. The inflammation characteristic of SLE can also damage the nervous system, and may result in abnormal sensation and weakness in the limbs (peripheral neuropathy); seizures; stroke; and difficulty processing, learning, and remembering information (cognitive impairment). Anxiety and depression are also common in SLE.
Scientists don’t know for certain what causes systemic lupus erythematosus (SLE). As an autoimmune disease, lupus occurs when your immune system attacks healthy tissue in your body. Studies have found that certain factors about your health, your genetics or your environment where you live may trigger lupus. The cause of lupus in most cases, however, is unknown:
- Genetic factors: Having certain genetic mutations may make you more likely to have lupus. It appears that people with an inherited predisposition for lupus may develop the disease when they come into contact with something in the environment that can trigger lupus.
- Hormones: Reactions to certain hormones in your body especially estrogen (female sex hormone) may make you more likely to develop lupus.
- Environmental factors: Aspects about where you live — including how much sunlight or how many toxins you’re exposed to — might affect your lupus risk. Exposure to the sun may bring on lupus skin lesions or trigger an internal response in susceptible people.
- Infections. Having an infection can initiate lupus or cause a relapse in some people.
- Medications. Lupus can be triggered by certain types of blood pressure medications, anti-seizure medications and antibiotics. People who have drug-induced lupus usually get better when they stop taking the medication. Rarely, symptoms may persist even after the drug is stopped.
- Your health history: Smoking, your stress level and having certain other health conditions (like other autoimmune diseases) might trigger lupus.
Anyone can develop lupus, but some groups of people have a higher risk:
- Women, especially women between the ages of 15 and 44.
- Black people.
- Hispanic people.
- Asian people.
- Native Americans, Alaska Natives and First Nations people.
- Pacific Islanders.
- People with a biological parent who has lupus.
Risk factors leading to systemic lupus erythematosus (SLE) include:
- Genetic predisposition, including haplotype HLA-B8, HLA-DR3
- Exposure to sunlight ultraviolet (UV) radiation
- Viral infection, particularly Epstein-Barr virus (EBV)
- Hormones
- Toxins such as cigarette smoke. SLE is more prevalent and more severe in smokers. Smoking also reduces the effectiveness of antimalarials and other therapies.
- Drugs in drug-induced lupus erythematosus
- Emotional upset.
Systemic lupus erythematosus (SLE) affects women more than men, at a ratio of 9 to 1 2. SLE is diagnosed most often in women in the first to fourth decades 3, the so called ‘child‐bearing years’ 4. In men, diagnosis is most common after age 59 2. Most recent studies have confirmed that females have higher incidence and prevalence regardless of age or ethnic origin 5. Gender differences in the clinical presentation of SLE have been reported 6. Rees et al 5 noted differences based on ethnicity, reporting that for either gender, prevalence of SLE was highest among those of black ethnicity, with white ethnic groups reporting the lowest prevalence, and Asian and Hispanic groups an intermediate prevalence.
Systemic lupus erythematosus (SLE) flares vary from mild to serious. Most patients have times when the disease is active, followed by times when the disease is mostly quiet – referred to as a remission. Yet, there is much reason for hope. Improvements in treatment have greatly improved these patients’ quality of life and increased their lifespan.
Systemic lupus erythematosus (SLE) can be difficult to diagnose because its signs and symptoms often mimic those of other ailments. Systemic lupus erythematosus can affect so many different organ systems, its symptoms can come and go, and no 2 people have exactly the same form of the disease. However, the most distinctive sign of systemic lupus erythematosus (SLE) is a facial rash that resembles the wings of a butterfly unfolding across both cheeks (systemic lupus erythematosus butterfly rash), which occurs in many (90% of cases) but not all cases of lupus. No one test can diagnose systemic lupus erythematosus. The combination of blood tests, urinalysis, chest X-ray, or an electrocardiogram (ECG), signs and symptoms, and physical examination findings leads to the diagnosis of lupus.
Laboratory tests for systemic lupus erythematosus (SLE):
- Complete blood count. This test measures the number of red blood cells, white blood cells and platelets as well as the amount of hemoglobin, a protein in red blood cells. Results may indicate you have anemia, which commonly occurs in lupus. A low white blood cell or platelet count may occur in lupus as well.
- Erythrocyte sedimentation rate (ESR). This blood test determines the rate at which red blood cells settle to the bottom of a tube in an hour. A faster than normal rate may indicate a systemic disease, such as lupus. The sedimentation rate isn’t specific for any one disease. It may be elevated if you have lupus, an infection, another inflammatory condition or cancer.
- Kidney and liver assessment. Blood tests can assess how well your kidneys and liver are functioning. Lupus can affect these organs.
- Urinalysis. An examination of a sample of your urine may show an increased protein level or red blood cells in the urine, which may occur if lupus has affected your kidneys.
- Antinuclear antibody (ANA) test. A positive test for the presence of these antibodies — produced by your immune system — indicates a stimulated immune system. While most people with lupus have a positive antinuclear antibody (ANA) test, most people with a positive ANA do not have lupus. If you test positive for ANA , your doctor may advise more-specific antibody testing.
If your doctor suspects that lupus is affecting your lungs or heart, he or she may suggest:
- Chest X-ray. An image of your chest may reveal abnormal shadows that suggest fluid or inflammation in your lungs.
- Echocardiogram. This test uses sound waves to produce real-time images of your beating heart. It can check for problems with your valves and other portions of your heart.
Lupus can harm your kidneys in many different ways, and treatments can vary, depending on the type of damage that occurs. In some cases, it’s necessary to test a small sample of kidney tissue to determine what the best treatment might be. The sample can be obtained with a needle or through a small incision.
Skin biopsy is sometimes performed to confirm a diagnosis of lupus affecting the skin.
Even with a confirmed diagnosis of systemic lupus erythematosus, treatments vary as much as the disease itself. Treatments depend greatly on which organs are affected and how severe your symptoms are. Determining whether you should be treated and what medications to use requires a careful discussion of the benefits and risks with your doctor. In general, however, the following oral medications are frequently used for systemic lupus erythematosus:
- Anti-malarial drugs such as hydroxychloroquine, chloroquine, or quinacrine. Medications commonly used to treat malaria, such as hydroxychloroquine (Plaquenil), affect the immune system and can help decrease the risk of lupus flares. Side effects can include stomach upset and, very rarely, damage to the retina of the eye. Regular eye exams are recommended when taking these medications.
- Corticosteroids. Prednisone and other types of corticosteroids can counter the inflammation of lupus. High doses of steroids such as methylprednisolone (Medrol) are often used to control serious disease that involves the kidneys and brain. Side effects include weight gain, easy bruising, thinning bones, high blood pressure, diabetes and increased risk of infection. The risk of side effects increases with higher doses and longer term therapy.
- Nonsteroidal anti-inflammatory drugs (NSAIDs). Over-the-counter nonsteroidal anti-inflammatory drugs (NSAIDs), such as naproxen sodium (Aleve) and ibuprofen (Advil, Motrin IB, others), may be used to treat pain, swelling and fever associated with lupus. Stronger NSAIDs are available by prescription. Side effects of NSAIDs may include stomach bleeding, kidney problems and an increased risk of heart problems.
- Immunosuppressants. Drugs that suppress the immune system may be helpful in serious cases of lupus. Immune-suppressing medications include azathioprine (Imuran, Azasan), mycophenolate (Cellcept), methotrexate (Trexall, Xatmep, others), cyclosporine (Sandimmune, Neoral, Gengraf) and leflunomide (Arava). Potential side effects may include an increased risk of infection, liver damage, decreased fertility and an increased risk of cancer.
- Biological agents. A different type of medication, belimumab (Benlysta, a human monoclonal antibody that inhibits B-cell activating factor or B-lymphocyte stimulator) administered intravenously, also reduces lupus symptoms in some people. Side effects include nausea, diarrhea and infections. Rarely, worsening of depression can occur. Rituximab (Rituxan, Truxima, a monoclonal antibody that depletes B cells from the circulation) may be beneficial for some people in whom other medications haven’t helped. Rituximab was originally used to treat lymphoma but is increasingly used for the treatment of autoimmune diseases. Side effects include allergic reaction to the intravenous infusion and infections.
In clinical trials, voclosporin has been shown to be effective in treating lupus. Other potential drugs to treat lupus are currently being studied, including abatacept (Orencia), anifrolumab and others.
You might need other medications or treatments to manage specific lupus symptoms you have or other health conditions it’s causing. For example, you may need treatment for anemia, high blood pressure (hypertension) or osteoporosis if lupus causes those issues.
Rheumatoid Arthritis
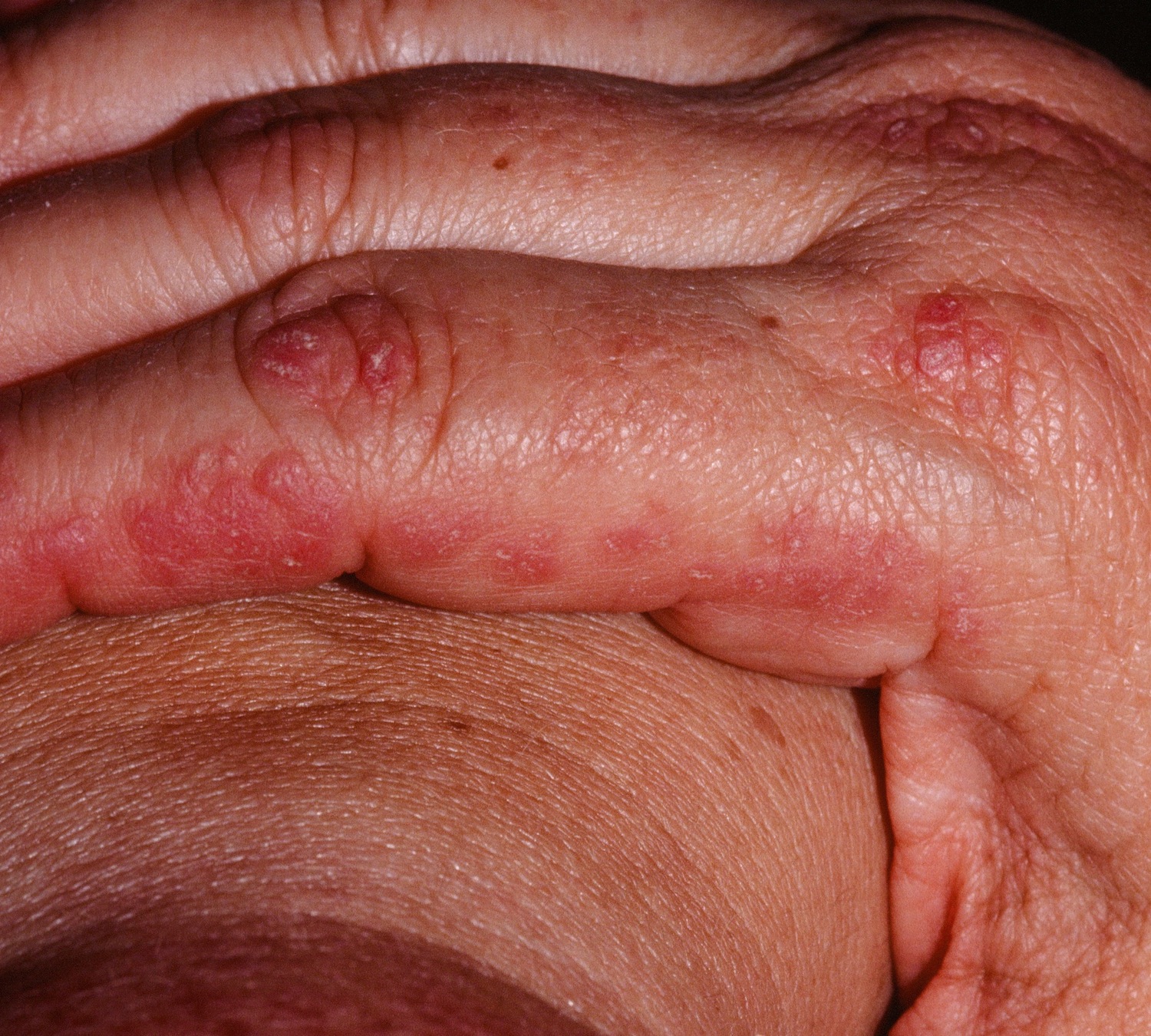
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory condition in which your immune system attacks the membrane lining that covers the ends of the bones in synovial joints (joint cavity that contains synovial fluid secreted by the synovial membrane or synovium which lines the articular capsule) for unknown reasons 7, 8, 9, 10. Rheumatoid arthritis (RA) causes painful, swollen, warm and stiff joints and loss of function in joints. Rheumatoid arthritis can affect the joints in your wrists, hands, elbows, shoulders, feet, spine, knees, and jaw. Rheumatoid arthritis often occurs in a symmetrical pattern, the inflammation typically affects the same joints on both sides of the body, meaning that if one knee or hand has rheumatoid arthritis, the other hand or knee is often also affected. Rheumatoid arthritis (RA) is usually first noticed in the small joints in the middle of the fingers and at the base of the fingers and toes, and sometimes in the elbows, ankles, or knees as well. Joints close to the torso, such as the shoulder joints or collarbone, may also become inflamed.
Rheumatoid arthritis is a chronic autoimmune inflammatory disease in which your body’s own immune system attacks the lining of the synovial membranes that surround the joints. Scientists don’t know the cause of rheumatoid arthritis. But it’s a condition in which the immune system attacks healthy joint tissue by mistake, called autoimmune disease. The cause is likely a mix of genetic changes and environmental factors from outside the body 11, 12. Hormones may play a role. There are also theories about certain viruses or bacteria causing autoimmune responses in people whose genes make them more likely to get it. Abnormal protein citrullination and the formation of anti-cyclic citrullinated peptide (anti-CCP) antibodies are critical pathogenic mechanisms in rheumatoid arthritis and are associated with severe joint lesions and extra-articular organ damage 13, 14, 15.
Figure 3. Rheumatoid arthritis
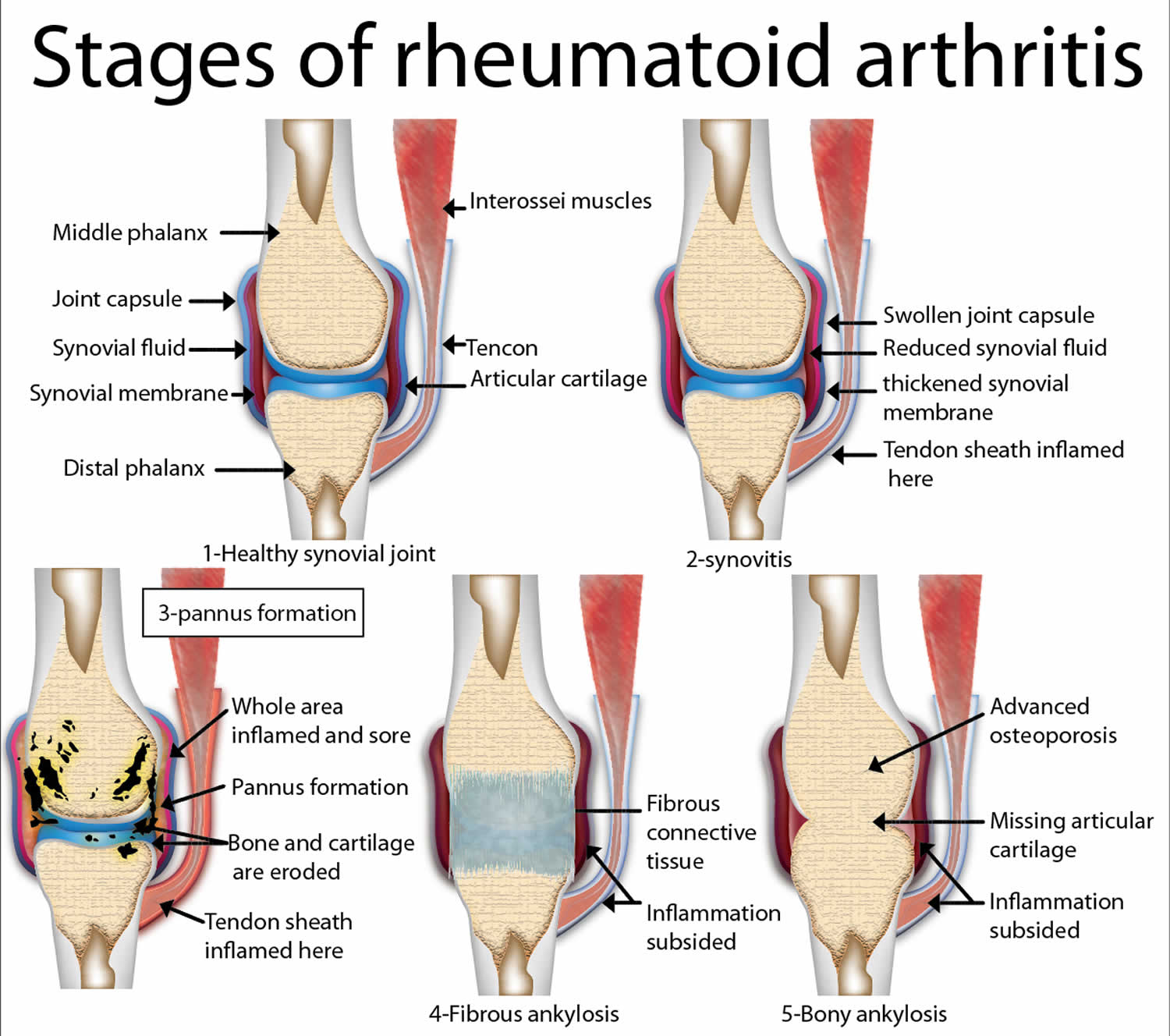
Figure 4. Rheumatoid arthritis
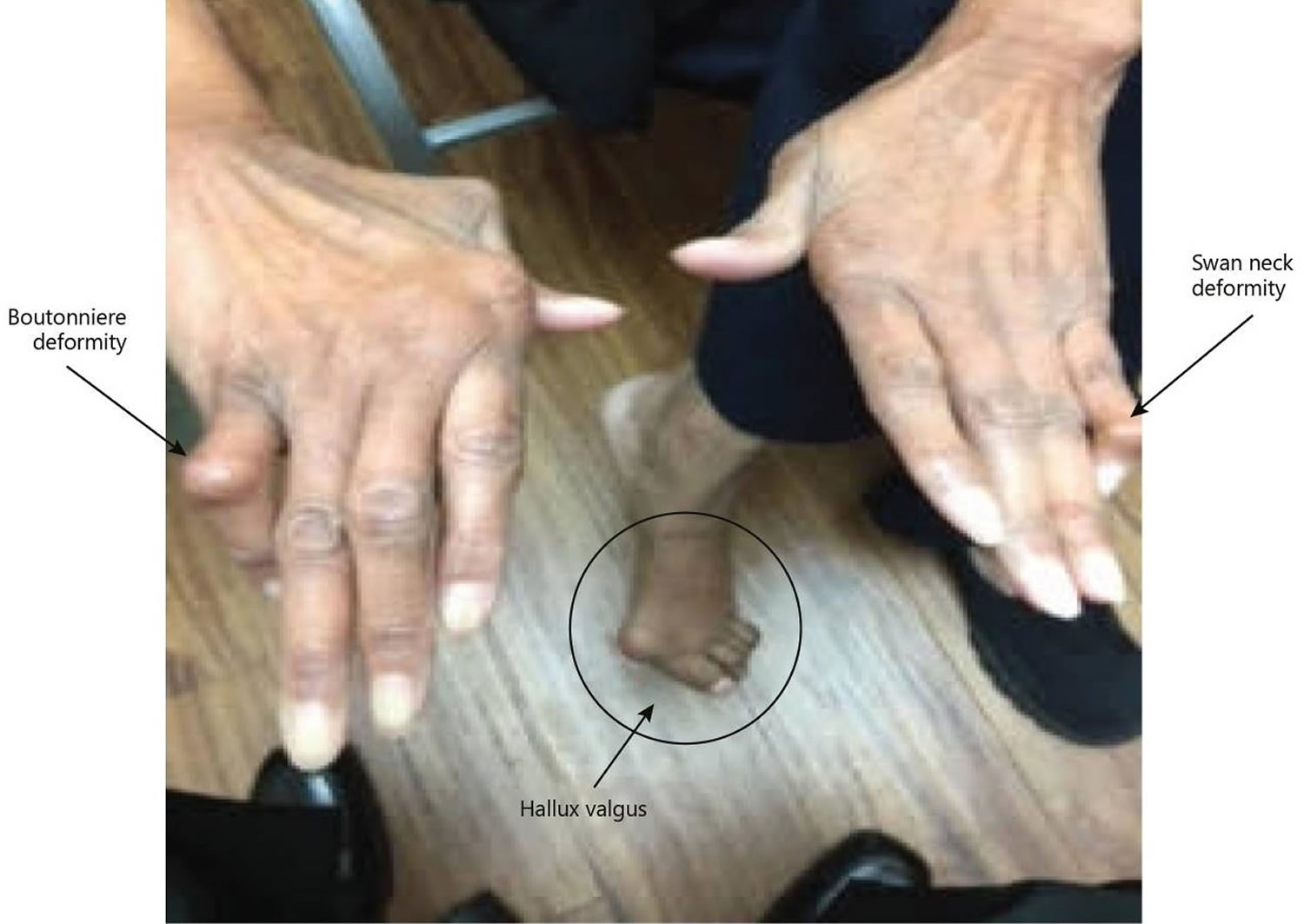
Footnote: A classic example of joint deformities associated with rheumatoid arthritis. Boutonniere deformity is visible in the 5th digit of the right hand, Swan neck deformity in the 5th digit of the left hand, and hallux valgus can be seen in the foot.
[Source 16 ]You are more likely to get rheumatoid arthritis (RA) if you have certain risk factors that include 17, 18, 19, 20, 21, 11, 22, 17, 23, 24:
- Age. Rheumatoid arthritis can happen at any age; however, the risk for developing rheumatoid arthritis increases with older age often begins in middle age from 30 to 50 years of age. Children and younger teenagers may be diagnosed with juvenile idiopathic arthritis (a group of autoimmune diseases causing joint inflammation in children and teenagers under 16 years old), a condition related to rheumatoid arthritis.
- Sex. Rheumatoid arthritis is more common among women than men approximately 70–80% of rheumatoid arthritis cases. Women are 2 to 3 times more likely than men to get rheumatoid arthritis, and do so about ten years earlier on average. Rheumatoid arthritis (RA) is also often more severe in women than in men. Researchers think that reproductive and hormonal factors may play a role in the development of rheumatoid arthritis for some women.
- Family history and genetics. Having a family member with rheumatoid arthritis or other autoimmune conditions may raise your risk of the condition. There are several genetic factors that slightly increase the risk of getting rheumatoid arthritis.
- Smoking. Research shows that people who smoke cigarette over a long period of time are at an increased risk of getting rheumatoid arthritis. Smoking also seems to make the condition worse in people who keep smoking. For people who continue to smoke, rheumatoid arthritis may be more severe.
- Obesity or Excess weight. Some research shows that being obese or overweight may increase your risk for getting rheumatoid arthritis as well as limit how much the disease can be improved.
- Periodontitis (gum infection or periodontal disease). A serious gum infection or periodontal disease can damage the soft tissue around your teeth and raise youre risk of getting rheumatoid arthritis.
- Lung diseases. Diseases of the lungs and airways may also be associated with developing rheumatoid arthritis.
Typical symptoms of rheumatoid arthritis are 25:
- Warm, swollen and painful joints
- Stiff joints in the morning after you wake up. They usually only become more flexible again after more than an hour.
- Weakness: Painful, stiff joints often end up not getting as much use, which can cause the muscles to get weaker over time.
- Exhaustion: Rheumatoid arthritis affects the whole body, so it often causes tiredness and general physical weakness.
- Rheumatoid nodules: As the disease progresses, small firm lumps called rheumatoid nodules sometimes develop under the skin. They’re usually not sensitive to pressure or touch.
Additional features of rheumatoid arthritis can include the following 10, 25:
- Fatigue, mild fevers, and a loss of appetite.
- Rheumatoid arthritis may cause other medical problems outside of the joints, in areas such as your heart, lungs, blood, nerves, eyes, and skin.
Rheumatoid arthritis can increase your risk of getting other medical problems such as 26, 25:
- Osteoporosis. Rheumatoid arthritis itself, and some medicines used to treat it, can increase your risk of osteoporosis, a condition where bones become weak and brittle, increasing the risk of fractures. Osteoporosis weakens bones and makes them more likely to break.
- Rheumatoid nodules. Rheumatoid nodules that are firm lumps just below the skin most often form around pressure points, such as the hands and elbows. But these nodules can form anywhere in your body, including the heart and lungs.
- Dry eyes and mouth called secondary Sjogren’s syndrome (a disease primarily affecting the glands that produce tears and saliva, leading to dryness of the eyes and mouth). People who have rheumatoid arthritis are much more likely to get a condition that lowers the amount of moisture in the eyes and mouth.
- Infections. Rheumatoid arthritis and many of the medicines used to treat it can harm the immune system. This can lead to more infections.
- Anemia due to low red blood cell counts.
- Carpal tunnel syndrome. If rheumatoid arthritis affects the wrists, the swelling can press on the median nerve to the hand and fingers.
- Heart problems. Rheumatoid arthritis can raise the risk of hardened and blocked arteries. It also can raise the risk of swelling and irritation, called inflammation of the sac enclosing the heart (pericarditis).
- Lung disease. People with rheumatoid arthritis have a higher risk of swelling and irritation, called inflammation of lung tissues. This can cause scarring and lead to shortness of breath that gets worse over time.
- Lymphoma. Rheumatoid arthritis raises the risk of a group of blood cancers that happen in the lymph system. This is called lymphoma. People with rheumatoid arthritis may have a higher risk of other cancers, as well.
Symptoms may gradually worsen, or they might not change for a long time. Sometimes the symptoms come and go in episodes, so the inflammation and pain may suddenly get worse and then improve again after a while. During phases when the symptoms are more severe, or at more advanced stages of the disease, people might sometimes feel extremely exhausted. This is known as “fatigue”.
Rheumatoid arthritis can progress in very different ways. In one study involving people with rheumatoid arthritis, ten years after they had developed the condition 25:
- Just under 50% of the participants reported minor limitations,
- A good 40% reported moderate limitations, and
- About 10% reported severe limitations in their everyday life.
These limitations include difficulties with things like getting up in the morning, getting dressed or preparing food – for example, opening packages, bottles or jars.
The late stages of rheumatoid arthritis can lead to major joint damage. Especially the joints in the hands can become very deformed, weak and stiff.
The mortality rate is 2.5 times higher in rheumatoid arthritis patients in comparison to healthy control 7. Management in rheumatoid arthritis patients increased their survival rate. However, rheumatoid arthritis patients have a lifespan of 5 to 10 years lower than healthy people 27.
Rheumatoid arthritis can be hard to diagnose in its early stages. That’s because the early symptoms can be like those of other common conditions. When trying to find out if you have rheumatoid arthritis, your doctor will first ask about your symptoms such as painful joints, stiff joints in the morning and general symptoms like tiredness or exhaustion (fatigue). During the physical exam, your doctor checks your joints for swelling, redness and warmth. Your doctor may also check your reflexes and muscle strength.
There’s no single test for rheumatoid arthritis. Your doctor may refer you to a rheumatologist (a doctor who specializes in arthritis care) for tests, diagnosis, and care.
Your doctor may order these tests:
- Blood tests: Blood tests are used to detect certain antibodies and signs of inflammation in the body. People with rheumatoid arthritis (RA) often have an elevated erythrocyte sedimentation rate (ESR), also called sed rate, or C-reactive protein (CRP) level. This may show a higher level of inflammation in the body. Other blood tests look for rheumatoid factor (RF) and anti-cyclic citrullinated peptide (anti-CCP) antibodies. Rheumatoid factor (RF) is an autoantibody that primarily targets the Fc fragment of IgG antibodies 28. Seropositive rheumatoid arthritis refers to rheumatoid arthritis where patients exhibit the presence of Rheumatoid factor (RF) or anti-cyclic citrullinated peptide (anti-CCP) antibodies, often involving early damage to lung tissue 29, 30. Anti-CCP antibodies and rheumatoid factor (RF) are two extensively utilized biomarkers in the diagnosis and prognosis of rheumatoid arthritis. Anti-MCV antibodies also known as anti-Sa antibodies have been proposed as an alternative due to their high specificity, which often outperforms anti-CCP antibody testing; nevertheless, it is unclear whether their benefits are significant enough to warrant routine clinical testing 24. The quest for novel biomarkers with significant therapeutic applications and their significance in rheumatoid arthritis (RA) remain a hot research area.
- Rheumatoid factor (RF) is detectable in about 60% of people diagnosed with rheumatoid arthritis (sensitivity); similarly, only about 60% specific, also occurring in older individuals, other immune mediated diseases, and in the context of infection 31. Typically pentameric IgM autoantibodies that bind the Fc portion of IgG (although can also occur in IgG and IgA isoforms). Rheumatoid factor (RF) likely has a role in perpetuating disease via immune complex formation and complement activation, leading to increased vascular permeability and immune cell chemotaxis to the joint.
- Anti-cyclic citrullinated peptide antibodies (ACPAs) is measured in routine practice using anti-cyclic citrullinated peptide assays. Anti-cyclic citrullinated peptide antibodies (ACPAs) are present in 60-80% of patients with rheumatoid arthritis 31. Anti-cyclic citrullinated peptide antibodies (ACPAs) are >90% specific in the setting of suspected rheumatoid arthritis (less specific at low titers and in the general, asymptomatic population) 31. Citrullination is a ubiquitous biochemical process catalyzed by the enzyme peptidyl arginine deiminase, leading to the post-translational modification of arginine amino acids; the presence of citrullinated auto-antigen is not associated with pathology, but the presence of anti-cyclic citrullinated peptide antibodies (ACPAs) is.
- Antiperinuclear factor (APF) and antikeratin antibodies (AKA). Antikeratin antibodies (AKA) and antiperinuclear factor (APF) have both demonstrated specificity for the diagnosis of rheumatoid arthritis 30. Antiperinuclear factor (APF) is found in 49 to 91% of rheumatoid arthritis patients, with a sensitivity range of 73 to 99%. Antikeratin antibodies are also seen in rheumatoid arthritis patients, with positive results occurring in 36 to 59% of cases 24, 32. Antikeratin antibodies (AKA) and antiperinuclear factor (APF) levels stay stable independent of disease duration. Antikeratin antibodies (AKA) and antiperinuclear factor (APF) can appear as early as the first stages of rheumatoid arthritis 30, 33. As a result, antikeratin antibodies (AKA) and antiperinuclear factor (APF) may aid in the early identification of rheumatoid arthritis, allowing for prompt intervention and drug delivery. These antibodies exhibit correlations with the presence of rheumatoid factor (RF), the activity and severity of rheumatoid arthritis, and with each other 30. Additional research has shown the utility of assessing anti-cyclic citrullinated peptide antibodies (ACPAs), antikeratin antibodies (AKA), antiperinuclear factor (APF), and certain rheumatoid factor (RF) isotypes for predicting early structural damage in the illness course 34. The best rheumatoid arthritis diagnostic marker among antikeratin antibodies (AKA), anti-CCP, and antiperinuclear factor (APF) remains unknown. According to a published study, antikeratin antibodies (AKA) and anti-CCP are both more effective than antiperinuclear factor (APF) in rheumatoid arthritis diagnosis. As a result, clinicians may choose antikeratin antibodies (AKA) or anti-CCP testing to aid with the diagnosis of rheumatoid arthritis 35.
- Anti-modified protein autoantibodies (AMPAs) aside from anti-cyclic citrullinated peptide antibodies (ACPAs), autoantibodies to carbamylated and acetylated protein antibodies (AMPAs) are well described and associated with rheumatoid arthritis; being unlikely to add diagnostic value, they are not routinely tested for, but remain of pathophysiological interest 31.
- Anti-mutated citrullinated vimentin (anti-MCV) also known as anti-Sa antibodies. Anti-mutated citrullinated vimentin (anti-MCV) is an important auto-antigen found in synovial tissue 24. In the diagnosis of rheumatoid arthritis, anti-Sa antibodies or anti-mutated citrullinated vimentin (anti-MCV) have a sensitivity of 20% to 40% and a specificity of 98% 24. Furthermore, anti-Sa antibodies or anti-mutated citrullinated vimentin (anti-MCV) strongly predict major joint issues and extra-articular rheumatoid arthritis symptoms 36. A new anti-MCV ELISA test was recently released, representing a step forward in rheumatoid arthritis diagnosis. According to Bang et al. 37, this test has the same specificity and sensitivity as anti-CCP antibodies. A published study evaluated the baseline antibodies against mutated citrullinated vimentin (MCV), CCP types 2 and 3 (both IgG isotype), and 3.1 (both IgG and IgA isotype) in 210 early rheumatoid arthritis patients over a two-year period. The Larsen score was used to assess disease activity at baseline and monthly during 24 months using radiographs of the hands and feet. Anti-MCV antibodies were associated with more severe illness, as seen by higher DAS28, ESR, and IgG levels, as compared to anti-CCP2, CCP3, and CCP3.1 antibodies 38. Another study looked at anti-MCV antibody levels at the start of treatment, one year later, and two years later in 162 individuals with early arthritis. When the indicated limit of 20 U/mL was used, the results showed that anti-MCV antibodies exhibited 92.3% specificity and 59.3% sensitivity. Patients with positive anti-MCV outcomes had higher Sharp-van der Heijde scores, higher ESR levels, and higher CRP levels at all assessment time points. 105 According to the published studies, anti-MCV antibodies may be a more sensitive and specific alternative to anti-CCP testing in diagnosing rheumatoid arthritis 39, 40.
- Antip68. Antibodies against the stress protein immunoglobulin heavy-chain binding protein (anti-BiP or antip68) are found in more than 60% of rheumatoid arthritis patients 24. These antibodies have also been found in experimental arthritis animal models 41. Furthermore, human stress protein BiP (immunoglobulin binding protein) levels in the rheumatoid joint are raised in sera from people in the early and pre-disease stages of rheumatoid arthritis 42. These data showed that human stress protein BiP (immunoglobulin binding protein) may be a significant auto-antigen in rheumatoid arthritis, but more study is needed to assess its usefulness as a biomarker.
- Other blood tests. Your doctor may also use other tests to check your kidney function, electrolytes, liver function, thyroid function, muscle markers, other autoimmune markers, and markers of infection to evaluate for your overall health and evaluate for other diagnoses. Other specific tests for rheumatoid arthritis, are sometimes considered.
- Imaging techniques: You may have X-rays to track rheumatoid arthritis in your joints over time. MRI scans and ultrasound tests may help with diagnosis. They can show how bad the condition is.
It can be difficult to diagnose rheumatoid arthritis at an early stage because the symptoms are often very mild in the first few weeks and months, and may not be typical. It is easier to diagnose rheumatoid arthritis (RA) in someone who has had it for a longer time. This is because, in addition to the typical physical symptoms, it’s often already easy to see changes in the joints.
If it’s thought that someone might have rheumatoid arthritis, specialized doctors known as rheumatologists can be consulted.
There is currently no cure for rheumatoid arthritis. Joint damage can happen quickly without treatment. But clinical studies show that easing of symptoms, called remission, is more likely with early treatment with medicines called disease-modifying antirheumatic drugs (DMARDs). Rheumatoid arthritis can also be treated with physical therapy and occupational therapy. There are also various support aids that can make some everyday tasks easier. People with rheumatoid arthritis are advised to do regular exercise or sports too.
Rheumatoid arthritis treatment options will depend on things like:
- how severe the inflammation and symptoms are,
- how far the disease has progressed,
- the predicted further course of the disease, and
- how well previous treatments have worked.
Your rheumatologist will suggest medicines based on how bad your symptoms are and how long you’ve had rheumatoid arthritis. You and your rheumatologist will decide on treatment. Medicines might include 31, 43:
- Nonsteroidal anti-inflammatory drugs (NSAIDs). Nonsteroidal anti-inflammatory drugs (NSAIDs) can relieve pain and ease swelling and irritation. NSAIDs you can get without a prescription include ibuprofen (Advil, Motrin IB, others) and naproxen sodium (Aleve). There also are stronger prescription NSAIDs. Side effects for all NSAIDs may include stomach upset, heart problems and kidney damage.
- Glucocorticoids. Corticosteroid medicines, such as prednisone (Rayos), ease inflammation and pain and slow joint damage. There can be serious side effects. The risk of side effects rises when taken at high doses over a long time. Side effects may include thinning of bones, fractures, easy bruising from skin thinning, weight gain, diabetes, cataracts and glaucoma, among others. Rheumatologists often prescribe a corticosteroid for quick symptom relief. The goal is to taper off the medicine when the condition is under control.
- Conventional disease-modifying antirheumatic drugs (DMARDs). These drugs can slow the progression of rheumatoid arthritis and save the joints and other tissues from long-term damage. Common disease-modifying antirheumatic drugs (DMARDs) include methotrexate (Trexall, Otrexup, others), leflunomide (Arava), hydroxychloroquine (Plaquenil, Sovuna) and sulfasalazine (Azulfidine). Side effects of disease-modifying antirheumatic drugs (DMARDs) vary but may include liver damage and severe lung infections.
- Biological DMARDs (bDMARDs) also known as biologic response modifiers, this newer class of DMARDs includes abatacept (Orencia), adalimumab (Humira), anakinra (Kineret), certolizumab (Cimzia), etanercept (Enbrel), golimumab (Simponi), infliximab (Remicade), rituximab (Rituxan), sarilumab (Kevzara) and tocilizumab (Actemra). Biologic DMARDs most often work best when used with a conventional DMARD, such as methotrexate. Biologic agents also raise the risk of rare infections such as tuberculosis, also called TB, or fungal infections. If you take biologic agents, you need to be watched closely.
- Targeted synthetic DMARDs (tsDMARDs). Your rheumatologist may prescribe these man-made medicines if conventional DMARDs and biological DMARDs (bDMARDs) haven’t worked. They include baricitinib (Olumiant), tofacitinib (Xeljanz) and upadacitinib (Rinvoq). Higher doses of tofacitinib may raise the risk of blood clots in the lungs, serious heart-related events and cancer.
Some common complementary and alternative treatments that have shown promise for rheumatoid arthritis include:
- Fish oil. Fish oils contain the omega−3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) that are known to reduce inflammation in the body and improve hypertriglyceridemia. Some studies have found that fish oil supplements may ease rheumatoid arthritis pain and stiffness. Side effects can include nausea, belching and a fishy taste in the mouth. Fish oil can get in the way of medicines you take. So check with your rheumatologist before trying it.
- Tai chi. This movement therapy involves gentle exercises and stretches and deep breathing. Many people use tai chi to relieve stress. Small studies have found that tai chi may improve mood and quality of life in people with rheumatoid arthritis. When led by a trained leader, tai chi is safe. But don’t do any moves that cause pain or make it worse.
Physical Therapy and Occupational Therapy
- Physical therapy and sports can help improve or maintain mobility, strength and joint function. Examples of suitable types of sports include cycling, brisk walking, dancing, doing exercises (e.g. gentle strengthening exercises), swimming and aqua aerobics.
- The main aim of occupational therapy is to maintain your mobility and hand strength, and to learn how to get by with rheumatoid arthritis in daily life.
- Assistive devices can make it easier to keep from stressing painful joints. For instance, a kitchen knife with a hand grip helps protect finger and wrist joints. Certain tools, such as buttonhooks, can make it easier to get dressed. Look for ideas in medical supply brochures and stores.
- In advanced arthritis, various aids can compensate for many physical limitations and help you to carry out everyday activities. These include orthopedic shoe inserts, grabbing aids and specially designed cutlery.
Psychological treatments can help relieve pain and minimize the impact it has on everyday life. They are also supposed to help relieve disease-related anxiety and depression that some people develop.
Surgery
Better drugs to treat rheumatoid arthritis have lowered the need for surgery. But if medicines fail to prevent or slow joint damage or your medications are not able to relieve your symptoms and the arthritis keeps getting worse, the thin layer that lines the joint (synovium) can be surgically removed. Surgery can reduce the pain and symptoms caused by severe joint damage resulting from rheumatoid arthritis. The type of surgery may depend on the joint involved. Your surgery may involve implanting an artificial joint or fusing the joint (arthrodesis), for example.
Scleroderma
Scleroderma also known as systemic sclerosis is an autoimmune disease where your immune system attacks healthy tissue affecting connective tissue (the tissue that connects joints, muscles, blood vessels and internal organs) in your body 44. In scleroderma, the connective tissue gets hard or thick. Scleroderma can cause swelling or pain in your muscles and joints. Scleroderma involves overproduction of a protein called collagen in connective tissue. This results in hardening of the skin (scleroderma means hard skin). The hallmark of scleroderma is hardening of the skin. Some forms of scleroderma can also damage your blood vessels and internal organs.
Scleroderma is not contagious, infectious, cancerous or malignant. Scleroderma has no cure, but symptoms and damage can be reduced with proper treatment.
It’s estimated that about 300,000 Americans have scleroderma. About one third of those people have the systemic form of scleroderma. Scleroderma affects women more often than men (female patients outnumber male patients about 4-to-1) and most commonly occurs between the ages of 30 and 50. However, scleroderma can develop in every age group from infants to the elderly.
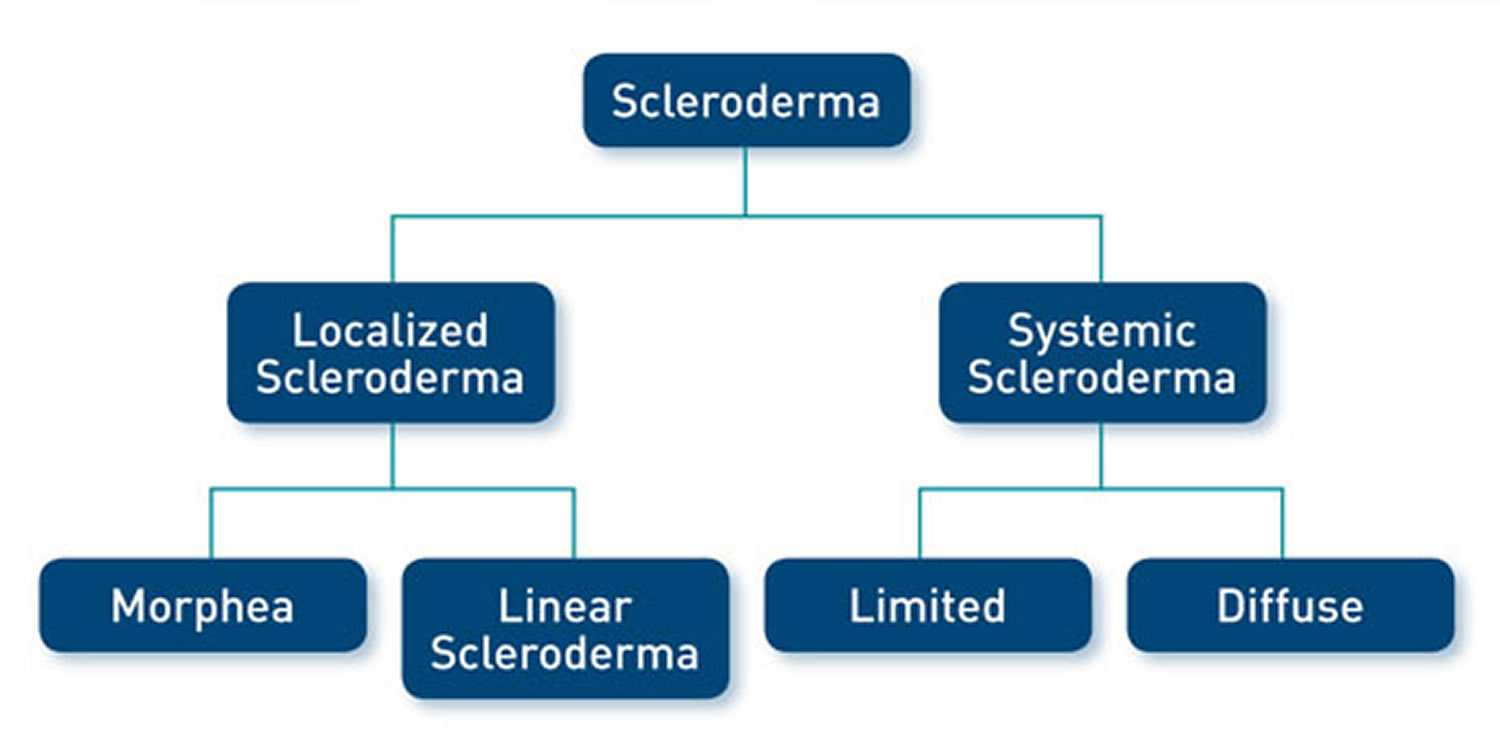
There are two types of scleroderma 44:
- Localized scleroderma also known as morphea, which only affects skin is more common in children. It does not harm major organs. It may get better or go away without help. But it can be severe in some people and can leave skin damage. Most localized scleroderma show up before age 40. They are also more common in people of European descent.
- Diffuse scleroderma also known as systemic sclerosis, which affects internal organs or blood vessels as well as skin. Systemic scleroderma are more common in people ages 30 to 50 and in African Americans.
The symptoms of scleroderma vary according to which part of your body is affected and it varies greatly for each person. Thickening and hardening of the skin is typical, especially on the fingers, arms and face. A mild scleroderma can become more serious if not properly treated. Prompt and proper diagnosis and treatment by qualified physicians may minimize the symptoms of scleroderma and lessen the chance for irreversible damage.
Another common symptom is Raynaud’s phenomenon, which is a blood circulation problem that causes your fingers or toes to change color and feel numb or painful in the cold. Primary Raynaud’s develops without any underlying condition whereas secondary Raynaud’s is linked to underlying disease, such as scleroderma.
Other symptoms of scleroderma include joint pain and stiffness, fatigue, indigestion or heartburn. Diffuse scleroderma can also cause symptoms related to the heart, lungs and kidneys.
There is currently no cure for scleroderma. However, treatment can improve symptoms. The treatment for scleroderma depends on what part of your body it is affecting. Your doctor may recommend stretching exercises for your joints, creams for your skin, dietary changes, or treatments to make the red patches on your skin less apparent.
Medication can improve blood circulation, and suppress the immune system, which may slow disease progress. If your organs are affected, you may be referred to a specialist, such as a kidney specialist if your kidneys are affected.
Lifestyle changes may make it easier to live with scleroderma. These include:
- wearing gloves and socks to keep your hands and feet warm, to prevent Raynaud’s phenomenon
- avoiding cigarette smoke, as this affects blood circulation
- regular physical activity to help keep skin and joints flexible
- keeping skin moisturized and clean to prevent dryness and infection
Figure 5. Scleroderma
Figure 6. Limited scleroderma (salt and pepper depigmentation in systemic scleroderma)
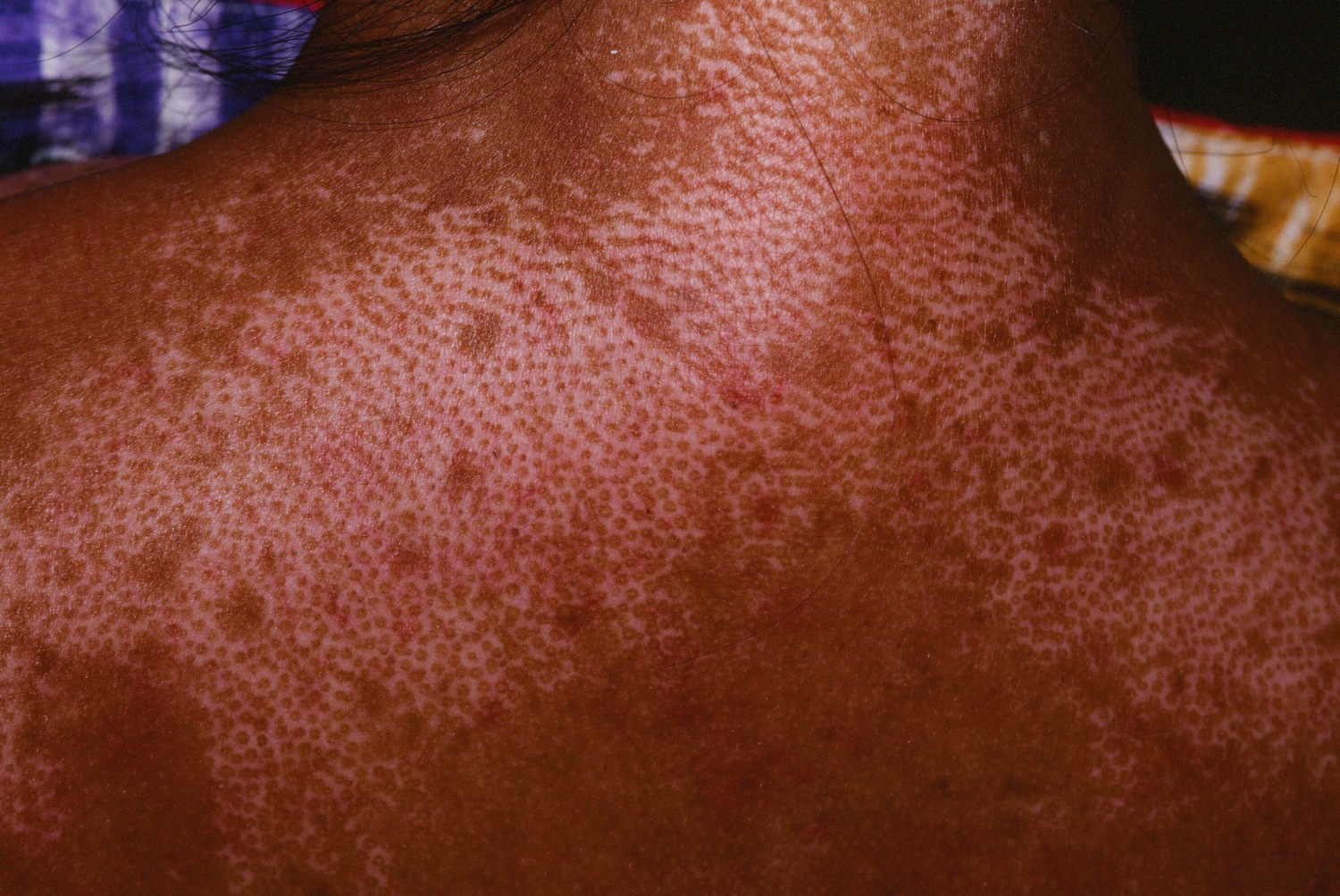
Figure 7. CREST scleroderma
Mixed Connective Tissue Disease (MCTD)
Mixed connective-tissue disease (MCTD) is a rare systemic autoimmune disease that is characterized by features commonly seen in at least two connective tissue diseases: systemic lupus erythematosus (SLE), scleroderma (systemic sclerosis), polymyositis (autoimmune disease that causes inflammation and weakness in the muscles), dermatomyositis (autoimmune disease that affects the skin and muscles, and sometimes the lungs), and rheumatoid arthritis (RA) with the presence of Raynaud’s disease also called Raynaud phenomenon (a condition where blood vessels in the fingers and toes constrict excessively in response to cold or stress, leading to reduced blood flow and color changes) and a distinctive antibody against what now is known to be U1-ribonucleoprotein (anti-RNP antibodies) 45, 46, 47, 48. The cause of mixed connective tissue disease is currently unknown.
Mixed connective-tissue disease (MCTD) was initially described by Sharp and colleagues in 1972 through a case series of 25 patients with features of systemic lupus erythematosus (SLE), scleroderma (systemic sclerosis), and inflammatory muscle disease (polymyositis) associated with anti-U1-ribonucleoprotein antibodies (anti-RNP antibodies) 47. However, at that time, mixed connective-tissue disease (MCTD) was not described as a separate entity from undifferentiated connective tissue disease, and its characteristics have evolved since then. Although most authors describe mixed connective-tissue disease (MCTD) as an independent entity, some believe it may represent an early stage of a definite connective tissue disease, such as systemic lupus erythematosus (SLE), scleroderma (systemic sclerosis), or the broader category of rheumatic “overlap syndromes”, a term used to describe when a patient has features of more than one classic inflammatory rheumatic disease. Mixed connective-tissue disease (MCTD) has no unique clinical features, and considerable interindividual variation in clinical signs and symptoms exists 45.
Multiple attempts have been made to develop classification criteria for mixed connective-tissue disease (MCTD), but there are currently no internationally agreed-upon diagnostic criteria for mixed connective-tissue disease (MCTD). Different classification and diagnostic criteria for mixed connective-tissue disease (MCTD) have been developed 49. These include the Alarcón-Segovia diagnostic criteria 50, 51 and, more recently in 2019, a set of criteria from a Japanese multispecialty consensus panel 52.
In 2019, a consensus panel in Japan proposed another revised set of diagnostic criteria for mixed connective-tissue disease (MCTD), which divides the disease features into 4 categories 52:
- The presence of a high titer of positive anti-U1-ribonucleoprotein antibodies (anti-RNP antibodies), and
- Raynaud phenomenon (Raynaud’s disease), swollen fingers or hand edema
- And either 1 of the following organ involvements or at least 2 overlapping signs and symptoms:
- Organ involvement includes:
- Pulmonary arterial hypertension (PAH)
- Aseptic meningitis
- Trigeminal neuropathy
- Overlapping signs and symptoms include:
- Systemic lupus erythematosus (SLE): polyarthritis, lymphadenopathy, malar rash, pericarditis or pleuritis (pleurisy), leukopenia or thrombocytopenia
- Scleroderma (systemic sclerosis): sclerodactyly, interstitial lung disease, esophageal dysmotility or dilatation
- Inflammatory myositis: muscle weakness, elevated levels of myogenic enzymes, myogenic abnormalities on electromyogram
- Organ involvement includes:
- Diagnosis is based on at least 1 common sign or symptom, presence of a high titer of positive anti-U1-ribonucleoprotein antibodies (anti-RNP antibodies), and either 1 characteristic organ involvement or at least 1 feature in 2 or more overlapping signs and symptoms. The Japanese diagnostic criteria have a sensitivity of 90.6% and a specificity of 98.4%, although they have not been formally adopted by the international community 53.
Today, most clinicians agree on a diagnosis of mixed connective-tissue disease (MCTD) if the following criteria are met 54, 50:
- The presence of a high titer of positive anti-U1-ribonucleoprotein antibodies (anti-RNP antibodies), and
- Raynaud phenomenon (Raynaud’s disease), swollen fingers or hand edema
- And at least 2 of the following:
- Synovitis
- Myositis
- Leukopenia
- Esophageal dysmotility
- Pleuritis
- Pericarditis
- Interstitial lung disease
Mixed connective-tissue disease (MCTD) has since been more completely characterized and is now recognized to consist of the following core clinical and laboratory features 55, 56, 57:
- Raynaud phenomenon (Raynaud’s disease)
- Swollen hands
- Arthritis or arthralgia
- Acrosclerosis
- Esophageal dysmotility
- Myositis
- Lung fibrosis 58
- Pulmonary hypertension
- High level of U1-ribonucleoprotein antibodies (anti-RNP antibodies)
- Antibodies against U1-70 kd small nuclear ribonucleoprotein (snRNP)
Despite the fact the diagnostic criteria for mixed connective-tissue disease (MCTD) have been agreed on, there continue to be debate amongst experts in rheumatology whether mixed connective-tissue disease (MCTD) is a distinct disease entity or should be considered a subset of lupus 59. A minority of authors continues to suggest that mixed connective-tissue disease (MCTD) would be better characterized as subgroups or early stages of disorders such as systemic lupus erythematosus (SLE) or scleroderma (systemic sclerosis) or an “overlap syndrome” (a term used to describe when a patient has features of more than one classic inflammatory rheumatic disease) 60. Other authors propose that mixed connective-tissue disease (MCTD) cases should not be distinguished from undifferentiated autoimmune rheumatic disease 61, 62.
In mixed connective-tissue disease (MCTD), the symptoms of the separate diseases usually don’t appear all at once. Instead, they tend to occur in sequence over a number of years, which can make diagnosis more complicated. Early signs and symptoms often involve the hands escpecially the fingers. Fingers might swell like sausages, and the fingertips become white and numb. In later stages, some organs — such as the lungs, heart and kidneys — may be affected.
Signs and symptoms of mixed connective tissue disease vary but may include Raynaud’s phenomenon; arthritis; heart, lung and skin abnormalities; kidney disease; muscle weakness, and dysfunction of the esophagus. The clinical signs, symptoms and manifestations of mixed connective tissue disease are similar among different ethnic groups. However, one study observed ethnic differences in the frequency of end-organ involvement; gastroesophageal reflux, sclerodactyly, and malar rash were significantly more common in a White group than in a group consisting of 57% Hispanics, 29% Blacks, and 14% Whites 63.
The onset of mixed connective tissue disease can occur anytime from early childhood to elderly adulthood, but typical age of onset is between 15-25 years old. The mean age of diagnosis was 48 years, and 84% of those affected were female 64. Mixed connective tissue disease is far more common in females than in males. Estimates of the female-to-male ratio vary from approximately 3:1 to 16:1 65, 66.
The point prevalence of mixed connective tissue disease has been found to be 3.8 per 100,000 adults in Norway, and is felt to be similar in many other parts of the world, though much higher prevalence of mixed connective tissue disease has been noted in some ethnic/geographic groups, notably in Japan. A population-based study from Olmsted County, Minnesota found that mixed connective tissue disease occurred in about 2 persons per 100,000 per year 67. Diagnosis was frequently delayed, with a median of 3.6 years elapsing from first symptom to fulfillment of diagnostic criteria 64. A study in American Indian and Alaska Native adults found a prevalence of 6.4 per 100,000 68. A nationwide study of mixed connective tissue disease in Norway found a point prevalence of 3.8 per 100,000 adults and an annual incidence rate of 2.1 per million 66. The prevalence of mixed connective tissue disease in Japan was estimated to be 2.7 per 100,000 65.
There is no cure for mixed connective tissue disease (MCTD) and due to the rarity of the condition, there are no randomized controlled trials to guide the treatment of patients with mixed connective-tissue disease 45. The overall goals of therapy for mixed connective tissue disease (MCTD) are to control symptoms, to maintain function, and to reduce the risk of future disease consequences 59.
The type of medication prescribed depends on the severity of your disease and your symptoms. Medications can include:
- Corticosteroids. Drugs, such as prednisone (Deltasone, Rayos), can help prevent your immune system from attacking healthy cells and suppressing inflammation. Side effects of corticosteroids can include mood swings, weight gain, high blood sugar, increased blood pressure, weakened bones and cataracts.
- Antimalarial drugs. Hydroxychloroquine (Plaquenil) can treat mild mixed connective tissue disease and might prevent flare-ups.
- Calcium channel blockers. Medications, such as nifedipine (Adalat CC, Procardia) and amlodipine (Norvasc), help relax the muscles in the walls of your blood vessels and may be used to treat Raynaud’s phenomenon.
- Other immunosuppressants. Your doctor might prescribe other medications based on your specific signs and symptoms. For example, if they’re similar to those of lupus, your doctor might recommend medications typically prescribed for people with lupus.
- Pulmonary hypertension medications. Bosentan (Tracleer) or sildenafil (Revatio, Viagra) may be prescribed. Pulmonary hypertension is typically less responsive to steroids, and the guidance of an expert in pulmonary hypertension should direct advanced management. Vasodilators such as prostaglandins, including epoprostenol; endothelin receptor antagonists, including ambrisentan; phosphodiesterase 5 inhibitors, including sildenafil; and immunosuppression with corticosteroids and cyclophosphamide may be appropriate therapeutic considerations.
Your doctor is likely to monitor you closely for signs of pulmonary hypertension.
The management of Raynaud phenomenon includes symptomatic strategies such as avoiding caffeine, smoking, cold temperatures, and injury to the digits. Oral calcium channel blockers, such as nifedipine, which decreases peripheral resistance, are an option. Prostaglandins, endothelin receptor antagonists, phosphodiesterase 5 inhibitors, and topical nitroglycerins are also effective.
Arthritis and arthralgia typically respond to nonsteroidal anti-inflammatory drugs (NSAIDs) and hydroxychloroquine (anti-malaria drug). For refractory synovitis, corticosteroids, methotrexate, and other disease-modifying anti-rheumatic drugs (DMARDs) can be used.
Pleuritis, pericarditis, myositis, and aseptic meningitis typically respond to steroids. Steroid-sparing agents, such as methotrexate, cyclosporine, azathioprine, and mycophenolate mofetil, are commonly used as second-line agents. Steroid-resistant myositis may respond to intravenous immunoglobulin.
Gastrointestinal reflux treatment involves proton pump inhibitors (PPI) or histamine blockers, lifestyle changes, and dietary modifications, such as elevating the head of the bed and avoiding dietary triggers. Prokinetics and gastric fundoplication are possible options for those who fail twice-daily proton pump inhibitor (PPI) therapy. Individuals with esophageal motility disorder may require prokinetics. Patients with malabsorption should be on a lactose-free diet, and medium-chain triglycerides should substitute for long-chain fatty acids.
Patients with autoimmune hemolytic anemia and thrombocytopenia are initially treated with steroids. Clinicians can consider rituximab in resistant cases.
Although, no controlled studies have been performed in mixed connective tissue disease, some patients with mixed connective tissue disease have been included in previous trials of lupus, scleroderma, myositis, and rheumatoid arthritis patients. In general, it appears that these mixed connective tissue disease subgroups respond similarly to treatments as have been reported in larger classical rheumatic disease-specific patient cohorts. These observations and accumulated clinical experience by mixed connective tissue disease experts supports the use of antimalarials for potential lupus-like disease modifying effects, the use of vasodilators to treat Raynaud’s phenomenon, the use of proton pump inhibitors for GERD (gastroesophageal reflux disease), and the use of additional disease-modifying anti-rheumatic drugs (DMARDs) for rheumatoid arthritis-like polyarthritis.
Cohort studies of mixed connective tissue disease patients with pulmonary hypertension or other lung disease have suggested that these patients may be more likely to respond well to a course of aggressive immunosuppression than is typical for patients with similar lung disease stemming from other causes.
Low-to-moderate doses of corticosteroids are often effective for rapid control of disease flares, and may be used as part of long-term therapy in some patients, despite their substantial long-term drug toxicities. Scleroderma renal crisis, a serious complication of scleroderma that is more likely after the use of high dose corticosteroids, has been infrequently reported in mixed connective tissue disease.
Nonsteroidal anti-inflammatory drugs (NSAIDs) may also be used to help control mild inflammatory symptoms, though their use must be balanced with their risk for gastrointestinal complications. NSAIDs rarely can cause aseptic meningitis in some individuals; this seems to occur slightly more often in patients with mixed connective tissue disease compared to other groups.
Figure 8. Raynaud’s disease fingers
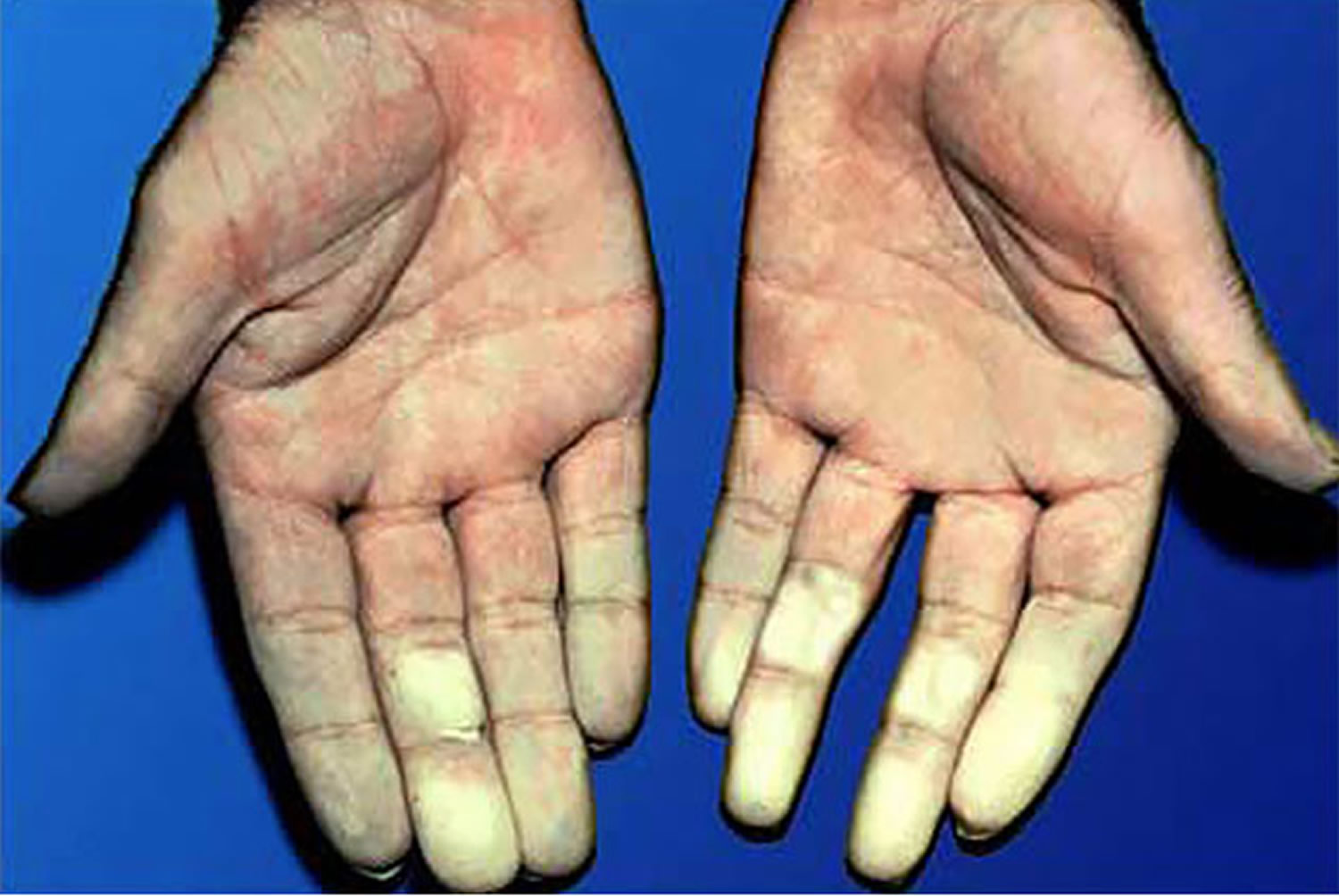
Figure 9. Scleroderma
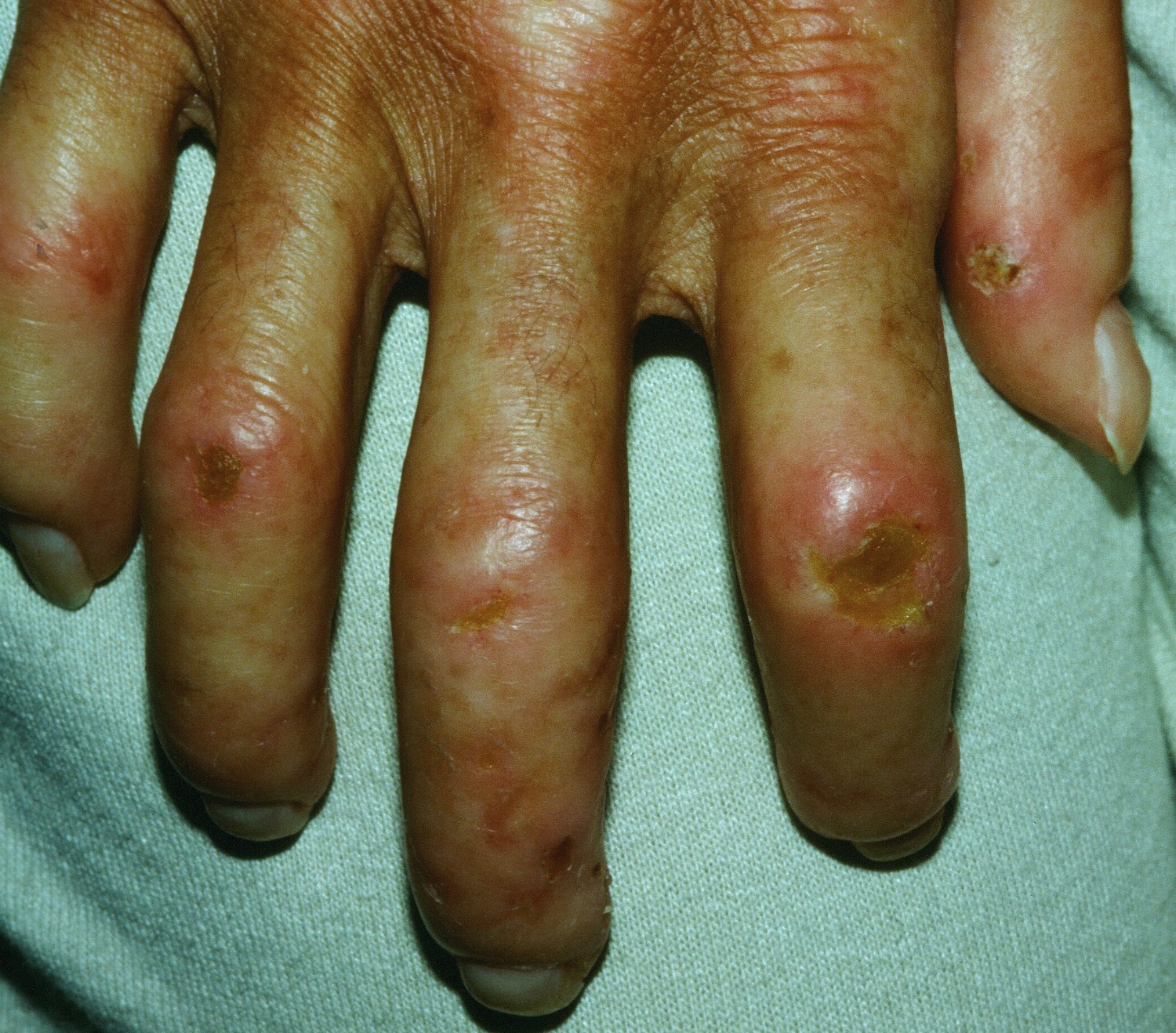
Figure 10. Polymyositis – mechanic’s hands
Relapsing polychondritis
Relapsing polychondritis is a rare autoimmune connective tissue disease which presents as severe, episodic, and progressive inflammation in cartilage bearing tissues like the ear, nose, larynx, trachea and bronchi, and may also involve the cardiovascular system, joints, eyes, skin, and kidneys 69, 70, 71, 72, 73, 74, 75, 76, 77. Ears, larynx and trachea may become “floppy” and the bridge of the nose can collapse into a “saddle nose” shape 78, 79. The aortic heart valve may also be affected. Relapsing polychondritis may also cause kidney inflammation and dysfunction. The inflammatory episodes are recurrent and unpredictable. A concomitant autoimmune disease also occurs in more than 30% of patients such as vasculitis 80, rheumatoid arthritis (RA) 81, and systemic lupus erythematosus (SLE) 82 or hematological diseases such as myelodysplastic syndrome (MDS) 83. Among Hungarian relapsing polychondritis patients a high prevalence (56%) of other autoimmune conditions was found, with Sjögren syndrome as the most common concomitant autoimmune disease 84, 85.
Jaksh-Wartenhorst described the first case with the name “polychondropathia” in 1923, when a 32-year-old patient presented with fever, pain, and swelling of the ears and later developed stenosis of external auditory canal and saddle nose deformity 86. A biopsy of the nasal cartilage showed the absence of cartilage. The term “relapsing polychondritis” was coined by Pearson and his coworkers in 1960 87. They recognized it as an inflammatory condition of the cartilaginous and noncartilaginous structures 87.
The cause of relapsing polychondritis is still not known 69, 88. Scientists suspect that relapsing polychondritis is an autoimmune condition 89. Autoimmune disorders also called autoimmune diseases are caused by the body’s immune system which normally helps protect your body from infection and disease begin attacking its own tissues (autoimmune) for unknown reasons 1. Some cases may be linked to abnormal reactions by blood cells (serum antibodies), to a thyroid protein (thyroglobulin), organ wall (parietal) cells, adrenal cells, or thyroid 89. Symptoms of relapsing polychondritis may arise when autoantibodies attack human cartilage.
Some researchers believe that relapsing relapsing polychondritis may be caused by an immunologic sensitivity to type II collagen, a normal substance found in skin and connective tissue.
Relapsing polychondritis is a rare autoimmune disease, being more common in Caucasians with the prevalence of 4.5 per million in a military population in the United States to 25 cases per million adults 90, 91. The annual incidence was estimated at 3.5 per million person-years in Rochester, MN, USA 92. However, a population-based cohort study conducted in the United Kingdom found a lower annual incidence, estimated at 0.71 per million person-years. The same study estimated the prevalence of relapsing polychondritis and estimated it at 9.0 cases per million population 93. The peak age at onset is between 40 years to 50 years, though it can occur at any age 94. Relapsing polychondritis occurs with equal frequency in both sexes and all racial groups. Over 30% of cases are associated with existing autoimmune condition or hematologic condition 95.
Clinical spectrum of relapsing polychondritis is variable and varies with duration of the disease and disease severity. Ear cartilage involvement is present in 90% of the cases, and inflammation is restricted to the cartilaginous portion of the ear with sparing of the ear lobes. Patients with relapsing polychondritis usually begin with the sudden onset of pain, tenderness, discoloration and swelling of the cartilage of one or both ears. The inflammation may spread to the fleshy portion of the outer ear causing it to narrow. Attacks may last several days to weeks before subsiding. Middle ear inflammation can cause obstruction of the eustachian tube. Recurrent attacks may lead to hearing loss.
Inflammation of both large and small joints is the second most common feature of relapsing polychondritis in 50% to 75% of patients. Wrist, metacarpophalangeal or the “knuckes” (the joint between the metacarpal head and the base of the proximal phalanx), proximal interphalangeal joints are commonly involved. Classic symptoms of pain and swelling are similar to those of arthritis.
Eye involvement occurs in 20% to 60% of relapsing polychondritis patients and involves episcleritis (inflammation of the episclera, the thin layer of tissue between the white of the eye [sclera] and the outer membrane [conjunctiva]), scleritis (inflammation of the sclera, the white outer layer of the eyeball, causing severe pain, redness with potential vision loss), keratitis (inflammation of the eye’s cornea, the clear tissue at the front of the eye that covers the pupil and iris), and uveitis (inflammation of the uvea, the middle layer of the eye containing blood vessels). Inflammation of the cartilage in the nose (nose chondritis) occurs in about 25% of cases. Nasal chondritis may be marked by cartilage collapse at the bridge of the nose resulting in a saddle nose deformity, nasal stuffiness or fullness and crusting.
In patients with relapsing polychondritis, up to 50% may experience airway involvement, and if unrecognized and left untreated it can lead to life threatening airway obstruction. Inflammation of the cartilage rings around trachea and bronchi results in the collapse of these airways (tracheobronchomalacia). Patient presents with a cough, speech difficulties, hoarseness of voice, wheezing and breathing difficulty. Respiratory tract involvement, specifically severe airway stenosis, can lead to acute respiratory failure, which is associated with a poor prognosis if not timely treated 96. Respiratory compromise is the most frequent cause of death in these patients.
Heart valve abnormalities, kidney inflammation and dysfunction also may occur.
The diagnosis of relapsing polychondritis is not always easy because there are no specific tests. Even when diagnosed, the treatment is not standardized. The drug treatment is tailored to each patient and the cornerstone of therapy is the use of glucocorticoids. For patients with severe relapsing polychondritis, other immunosuppressive are used including methotrexate and cyclophosphamide 97. Recently newer biological agents have also been used to manage relapsing polychondritis patients with varying results. However, prior to initiating treatment with these novel agents, the patient needs a thorough work up to ensure that he or she is fit to receive the therapy. Any patient with worsening of symptoms should be referred to the specialist 98. People who develop severe heart or respiratory complications may require surgery.
Figure 11. Relapsing polychondritis

Footnotes: A 31-year-old man presented to the emergency department with a 2-week history of swelling of the left ear and a 6-month history of weight loss, fatigue, and generalized aches. During the 2 years before presentation, he had been treated multiple times with antibiotics for recurrent episodes of pain in both ears. The physical examination revealed a tender, erythematous, and edematous left pinna, with sparing of the lobule (Panel A). The patient also had a prominent saddle-nose deformity that had developed over the previous year (Panel B). The costochondral joints were tender on palpation, and the left knee was swollen and tender. Laboratory studies showed an erythrocyte sedimentation rate of more than 120 mm per hour (reference range, 0 to 15). He received a diagnosis of relapsing polychondritis, a systemic autoimmune condition that affects cartilaginous structures, particularly of the ears, nose, joints, larynx, and large airways. Prednisone was prescribed at a dose of 40 mg daily, and the patient had some alleviation of the pain and swelling within 2 weeks. After 1 month, therapy with methotrexate was started, and the prednisone was slowly tapered over a period of 6 months.
[Source 99 ]Figure 12. Relapsing polychondritis anterior acleritis (inflammation of the episcleral and scleral tissues)
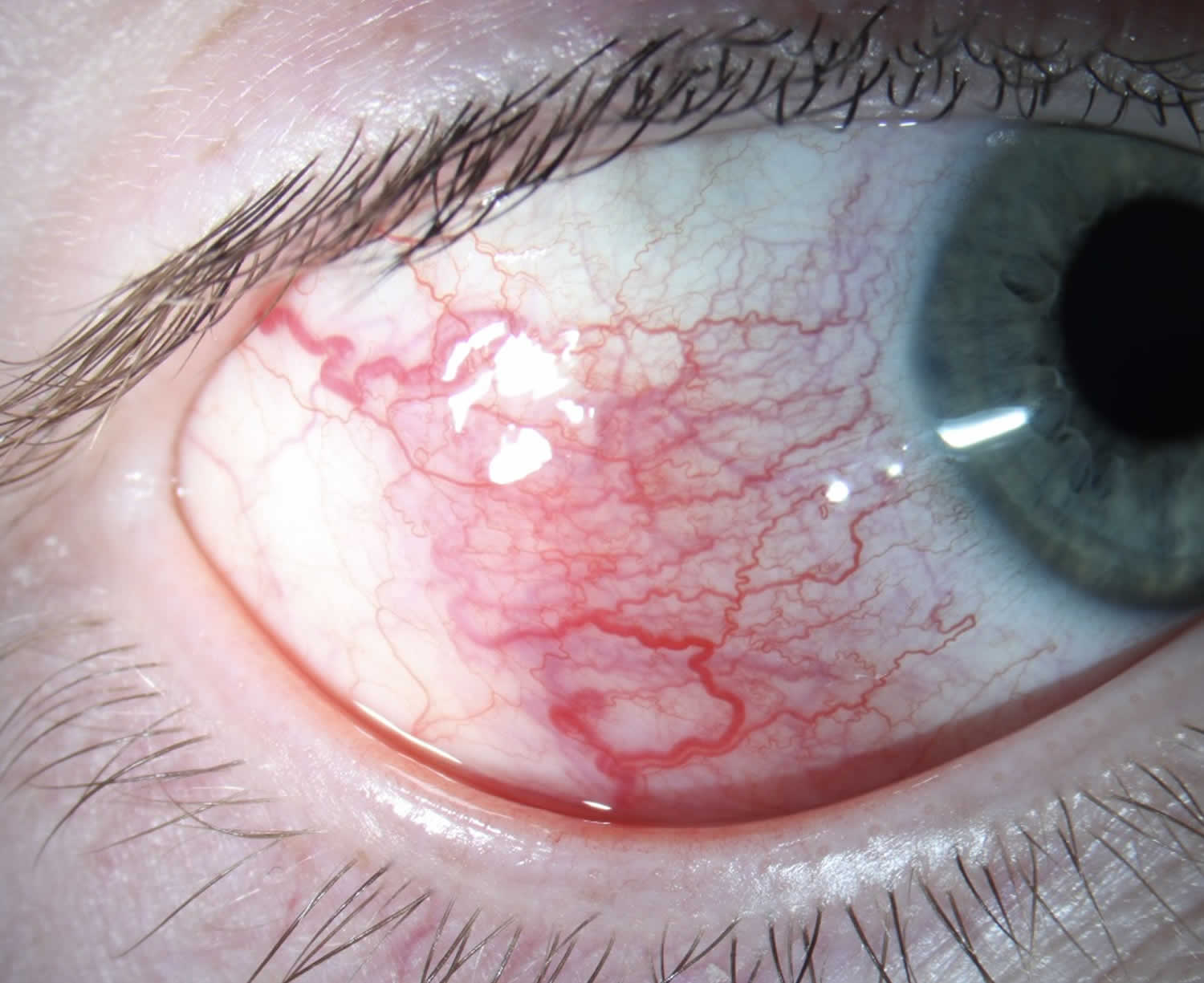
Footnotes: Anterior Scleritis is inflammation of the episcleral and scleral tissues that presents with severe injection of superficial episcleral and deep scleral vessels. Depending on the severity, anterior scleritis may be associated with eye complications including loss of visual acuity, anterior uveitis, peripheral ulcerative keratitis, glaucoma, and retinal or choroidal detachment, and frequently requires work-up for associated systemic inflammatory conditions such as rheumatoid arthritis or, as in this case, relapsing polychondritis. Therefore, it is important to differentiate it from benign or self-limiting episcleritis. On slit lamp exam, visible vessels have classically demonstrate a crisscrossing pattern with conjunctival vessels oriented radially and episcleral vessels oriented in a more circumferential pattern, and yield a characteristic “violaceous hue”. Unlike conjunctival vessels, episcleral or deeper scleral vessels cannot be moved with a cotton-tipped applicator. Subconjunctival edema may be present. Instillation of phenylephrine drops will blanch the superficial conjunctival vessels but not the deeper episcleral vessels.
[Source 100 ]Myositis
Myositis means inflammation of the muscles that you use to move your body. An injury, infection, or autoimmune disease can cause myositis. Two specific kinds of myositis are polymyositis and dermatomyositis. Polymyositis causes muscle weakness, usually in the muscles closest to the trunk of your body. Dermatomyositis causes muscle weakness, plus a skin rash.
Other symptoms of myositis may include:
- Fatigue after walking or standing
- Tripping or falling
- Trouble swallowing or breathing
Doctors may use a physical exam, lab tests, imaging tests and a muscle biopsy to diagnose myositis. There is no cure for these diseases, but you can treat the symptoms. Polymyositis and dermatomyositis are first treated with high doses of a corticosteroid. Other options include medications, physical therapy, exercise, heat therapy, assistive devices, and rest.
Sjögren’s syndrome
Sjogren’s syndrome also known as Sjögren’s disease is a long-lasting (chronic) autoimmune disorder that happens when your immune system attacks the glands that make tears (lacrimal glands) in your eyes, salivary glands that make saliva in your mouth, and other parts of your body (multi-organ problems) 101, 102, 103, 104, 105, 106, 107, 108, 109. The main symptoms of Sjögren’s syndrome are dry eyes (xerophthalmia) and dry mouth (xerostomia), but other parts of your body may be affected as well, with many people reporting chronic fatigue, fever and joint and muscle pain 110. With Sjogren’s syndrome you may have dryness in other places that need moisture, such as your nose, throat, and skin. In addition, Sjögren’s syndrome can also damage other parts of your body including your lungs, kidneys, blood vessels, digestive organs, vagina and nervous system 111. Most people with Sjogren’s syndrome are women, with women are 10 times more likely to have Sjogren’s syndrome than men 112, 113, 114.
The first clinical description of Sjögren syndrome was by Mikulicz in 1892, who described a 42-year-old with bilateral parotid and lacrimal gland enlargement. In 1933, the Swedish Ophthalmologist Henrik Sjögren compiled a clinical and histopathologic description of a series of patients with the “sicca complex” of dry eyes and mouth.
Doctors have two categories for Sjogren’s syndrome 108, 101, 115:
- Primary Sjogren’s syndrome: Occurs if you do not have other rheumatic diseases.
- Secondary Sjogren’s syndrome (Sjogren-overlap syndrome): Occurs if you already have another rheumatic disease, such as rheumatoid arthritis or systemic lupus erythematosus (SLE), scleroderma, or polymyositis.
The cause of Sjogren’s syndrome is unknown. Sjögren’s syndrome or Sjögren’s disease is an autoimmune disorder that happens when the immune system attacks and damages glands in your body that produce and control moisture. Normally, your immune system protects your body from infection and disease. Researchers theorize that Sjögren’s disease could be caused by complex interplay of genetic, hormonal, and environmental factors 101. The leading “pathogenic model” suggests that environmental insults in genetically predisposed individuals injure the salivary gland epithelium. This epithelial damage initiates inflammation and triggers an abnormal immune response, driving the chronic autoimmune process that defines Sjögren’s disease 116. A genetic study identified a strong correlation between Sjögren syndrome and the HLA-DQB1 gene variants 117. These major histocompatibility complex variants (changes or mutations) appear to promote an abnormal immune response when combined with specific environmental triggers 117. Laboratory findings and indirect epidemiological data implicate viral infections, particularly Epstein-Barr virus (EBV), in Sjögren’s disease development 118, 119. Exposure to solvents and inorganic chemicals has also been associated with the development of Sjögren syndrome 120.
Sjogren’s syndrome affects 400,000 to 4 million people (approximately 0.5% to 1.0% of the population) in the United States. Although Sjögren syndrome can develop at any age, symptoms most commonly begin between the ages of 45 and 55. Sjögren syndrome occurs globally in adults and less commonly in children across all racial and ethnic backgrounds. Approximately half of those diagnosed with Sjogren’s syndrome also have other autoimmune conditions such as rheumatoid arthritis or systemic lupus erythematosus (SLE) or Raynaud’s disease (Raynaud’s phenomenon). Most people with Sjogren’s syndrome are able to live normally, without any serious complications – especially if they take care to manage their symptoms.
The lack of an evidence-based, standardized screening tool to identify which dry eye patients require evaluation for Sjögren syndrome contributes to the underreferral of these individuals 101. As a result, many cases of Sjögren syndrome remain undiagnosed, perpetuating a pattern of underrecognition and delayed diagnosis 121.
The 2 main symptoms of Sjogren’s syndrome are:
- Dry eyes. Your eyes may burn or itch or feel like they have sand in them. Sometimes, the dryness causes blurry vision or sensitivity to bright light. You may get irritated, itchy eyelids due to inflammation.
- Dry mouth. Your mouth may feel chalky, and you may have trouble swallowing, speaking, and tasting. Because you lack the protective effects of saliva, you may develop more dental decay (cavities) and mouth infections, such as candidiasis (also called thrush).
Most individuals with Sjögren syndrome present with sicca symptoms, such as xerophthalmia (dry eyes), xerostomia (dry mouth), and parotid gland enlargement 122.
In some people, the main problem is dry mouth, while for others it is dry eyes, and some people experience both problems equally. In some cases, Sjögren’s disease affects other tissues and organs and has more widespread effects on the body.
Some people with Sjogren’s syndrome also have one or more of the following:
- Fatigue
- Joint pain, swelling and stiffness
- Dry skin
- Skin rashes
- Dry gritty eyes
- Dry nasal passages and throat
- Dry cough
- Decreased sense of taste and smell
- Dry mouth with difficulty swallowing or talking
- Sore tongue or throat
- Muscle aches and pain
- Muscle weakness
- Stomach upset, irritable bowel
- Acid reflux.
- Vaginal dryness.
- Swelling of the glands around the face and neck.
- Swollen salivary glands — particularly the set located behind your jaw and in front of your ears (parotid glands)
- Trouble sleeping.
- Poor concentration and memory problems.
- Numbness, tingling, and weakness, especially in the hands or feet (peripheral neuropathy)
- Shortness of breath or trouble breathing.
- Recurrent bronchitis or pneumonia.
Sjögren’s disease may have different effects on the body, and the symptoms vary from person to person. In some people, Sjögren syndrome symptoms cycle between mild and severe. Sjögren syndrome symptoms can be severe, with some people reporting debilitating pain and fatigue.
People with Sjögren syndrome have a higher chance of developing a type of cancer called lymphoma, but the risk of developing it is low.
To make a diagnosis, doctors may use a medical history, physical exam, certain eye and mouth tests, blood tests, and biopsies.
Sjogren’s syndrome is primarily managed by rheumatologists, in collaboration with ophthalmologists and oral medicine/pathology specialists.
There is no cure for Sjögren’s disease, but there are several ways to treat and manage the symptoms. Sjogren’s syndrome usually affects people differently so it is important to discuss treatment options with a health practitioner to decide on the best treatment plan.
Treatment can vary depending on the severity of your symptoms. Treatment is usually aimed at relieving your symptoms and preventing or minimizing complications and damage to tissues, such as the surface of the eye.
- Dry eyes can be treated with over-the-counter or prescription artificial tears, tear stimulants, and/or thicker eye lubricants. Sometimes plugs may be inserted into the drainage ducts in the eyes to help tears remain on the surface of the eyes.
- Dry mouth may be helped by frequent small drinks of water or sugarless chewing gum to stimulate saliva production, and mouth lubricants can be used as necessary. In some cases, medications that increase saliva production may be prescribed.
- Regular dental care and checkups are important as those affected are prone to cavities.
- Joint pain and other arthritis symptoms are treated with anti-inflammatory medicines, such as aspirin and other nonsteroidal anti-inflammaory drugs (NSAIDs).
- In some severe cases when internal organs of the body are affected, drugs that dampen the immune system (immunosuppressants) and steroids may be prescribed.
Figure 13. Sjogren’s syndrome rash (cutaneous vasculitis)
Figure 14. Sjogren’s syndrome parotid gland swelling
Inherited connective tissue disorders
Inherited connective tissue disorders also called heritable connective tissue disorders are a group of genetic conditions affecting the body’s connective tissues. Connective tissue provides the support and structure to other tissue and organs in the body. Inherited connective tissue disorders are multisystemic conditions, as such the presentation can vary between individuals, and individually across the lifespan. Due to the multisystem nature of inherited connective tissue disorders all organ systems can be impacted, this includes the skin, bones, joints, eyes, heart, and blood vessels.
Ehlers-Danlos syndrome
Ehlers-Danlos syndrome is a group of genetic collagen disorders caused by genetic changes that affect connective tissues supporting the skin, bones, blood vessels, and many other organs and tissues throughout your body 124. Defects in connective tissues cause the signs and symptoms of Ehlers-Danlos syndrome, which range from mildly loose joints (joint hypermobility), skin hyperextensibility, and tissue fragility to life-threatening complications such as arterial rupture, organ rupture, joint dislocation, chronic pain, and severe fatigue, among many others. Ehlers-Danlos syndrome symptoms and severity vary widely. People with Ehlers-Danlos syndrome may experience other symptoms, including intestinal and mental health issues, and postural orthostatic tachychardia syndrome (POTS), which causes a racing heart and dizziness on standing. These complications vary, depending on a person’s age, sex, lifestyle and the genes they inherit. Some complications might also result from co-existing conditions, poor nutrition or incorrect or inadequate treatment of the underlying disease.
The various forms of Ehlers-Danlos syndrome have been classified in several different ways. Originally, 11 forms of Ehlers-Danlos syndrome were named using Roman numerals to indicate the types (type I, type II, and so on). In 1997, researchers proposed a simpler classification, the Villefranche nomenclature, that reduced the number of types to six and gave them descriptive names based on their major features. In 2017, the classification was updated to include rare forms of Ehlers-Danlos syndrome that were discovered more recently. The 2017 classification describes 13 types of Ehlers-Danlos syndrome 125.
The most common types of Ehlers-Danlos syndrome are hypermobile Ehlers-Danlos syndrome (hEDS) and classical Ehlers-Danlos syndrome (cEDS) that have extremely flexible joints which are prone to frequent dislocations and stretchy, fragile skin. As many as half of people with hypermobile Ehlers-Danlos syndrome may have postural orthostatic tachychardia syndrome (POTS). People who have Ehlers-Danlos syndrome can get deep wounds from minor injuries, but their skin may not be strong enough for stitches and often forms conspicuous scars. The most severe subtype, vascular Ehlers-Danlos syndrome, can cause fatal ruptures of blood vessels, the intestine and uterus.
The combined prevalence of all types of Ehlers-Danlos syndrome appears to be at least 1 in 5,000 individuals worldwide. It affects both men and women with no predisposition to race or ethnicity. The hypermobile and classical forms are most common; the hypermobile type may affect as many as 1 in 5,000 to 20,000 people, while the classical type probably occurs in 1 in 20,000 to 40,000 people. Other forms of Ehlers-Danlos syndrome are rare, often with only a few cases or affected families described in the medical literature. There may be a family history – for instance in vascular Ehlers-Danlos syndrome sudden death in a close relative. Progress on the Human Genome Project has provided valuable information regarding the actual genes involved. Ehlers-Danlos syndrome is usually diagnosed in younger patients as typical features such as joint laxity, skin fragility and scarring tendencies are recognisable from early childhood.
An unusually large range of joint movement (hypermobility) occurs in most forms of Ehlers-Danlos syndrome, and it is a hallmark feature of the hypermobile type. Infants and children with hypermobility often have weak muscle tone (hypotonia), which can delay the development of motor skills such as sitting, standing, and walking. The loose joints are unstable and prone to dislocation, chronic pain and early-onset arthritis. In the arthrochalasia type of Ehlers-Danlos syndrome, infants have hypermobility and dislocations of both hips at birth.
Many people with the Ehlers-Danlos syndromes have soft, velvety skin that is highly stretchy (elastic) and fragile. Affected individuals tend to bruise easily, and some types of the condition also cause abnormal scarring. People with the classical form of Ehlers-Danlos syndrome experience wounds that split open with little bleeding and leave scars that widen over time to create characteristic “cigarette paper” scars. The dermatosparaxis type of the disorder is characterized by loose skin that sags and wrinkles, and extra (redundant) folds of skin may be present.
Some forms of Ehlers-Danlos syndrome, notably the vascular type and to a lesser extent the kyphoscoliotic, classical, and classical-like types, can cause unpredictable tearing (rupture) of blood vessels, leading to internal bleeding and other potentially life-threatening complications. The vascular type of Ehlers-Danlos syndrome is also associated with an increased risk of organ rupture, including tearing of the intestine and rupture of the uterus during pregnancy.
Other types of Ehlers-Danlos syndrome have additional signs and symptoms. The cardiac-valvular type causes severe problems with the valves that control the movement of blood through the heart. People with the kyphoscoliotic type experience severe curvature of the spine that worsens over time and can interfere with breathing by restricting lung expansion. A type of Ehlers-Danlos syndrome called brittle cornea syndrome is characterized by thinness of the clear covering of the eye (the cornea) and other eye abnormalities. The spondylodysplastic type features short stature and skeletal abnormalities such as abnormally curved (bowed) limbs. Abnormalities of muscles, including hypotonia and permanently bent joints (contractures), are among the characteristic signs of the musculocontractural and myopathic forms of Ehlers-Danlos syndrome. The periodontal type causes abnormalities of the teeth and gums.
If you have Ehlers-Danlos syndrome, you will need to be careful when doing activities that can put stress on your joints and increase the risk of injury, such as contact sports.
There is no specific treatment for Ehlers-Danlos syndrome, but symptoms can be managed through:
- medications to ease paid and reduce blood pressure
- physical therapy such as exercises or physical braces to keep joints as stable and strong as possible.
If you have Ehlers-Danlos syndrome, you may see a range of health professionals to help you manage your condition, including physiotherapists, occupational therapists, psychologists, rheumatologists and genetic counselors.
Figure 15. Ehlers–Danlos syndrome
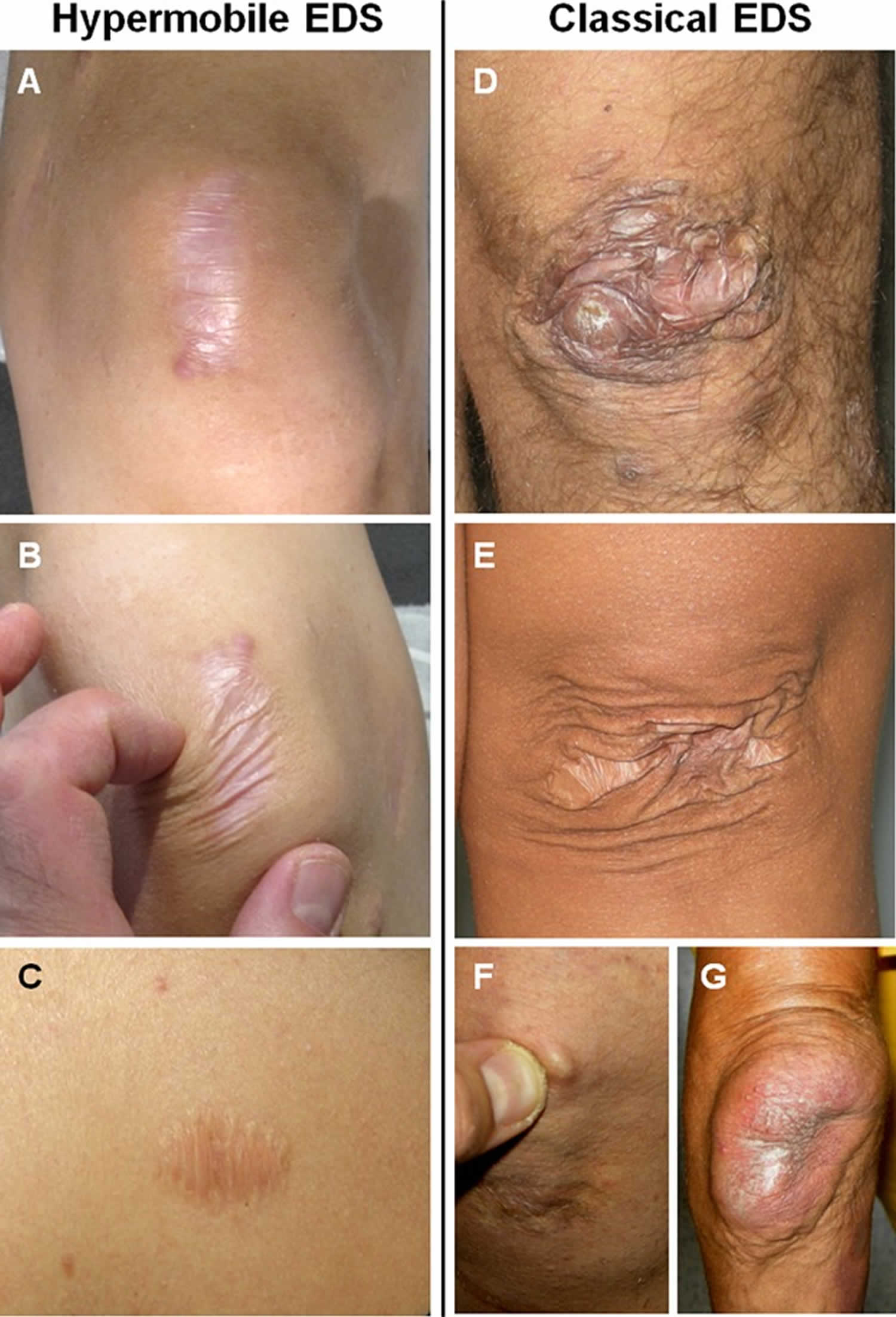
Footnotes: Hypermobile Ehlers-Danlos syndrome (hEDS) images A to C. Post-traumatic, atrophic, and widened scar in a young man (A). Skin stretching between the examiner’s fingers discloses mild atrophy of the underlying dermis (B). A further atrophic and widened scar due to wound healing delay after excision of a melanocytic nevus in a young woman (C). Classical Ehlers-Danlos syndrome (cEDS) images D to G. Typical papyraceous and hemosideric scar after repetitive wound re-opening and molluscoid pseudotumor in an adult man (D). Papyraceous, but not hemosideric scar and acquired cutis laxa in a young woman (E). Subcutaneous spheroid (F). Huge molluscoid pseudotumor of the elbow (G).
[Source 125 ]Table 1. Ehlers Danlos syndrome types
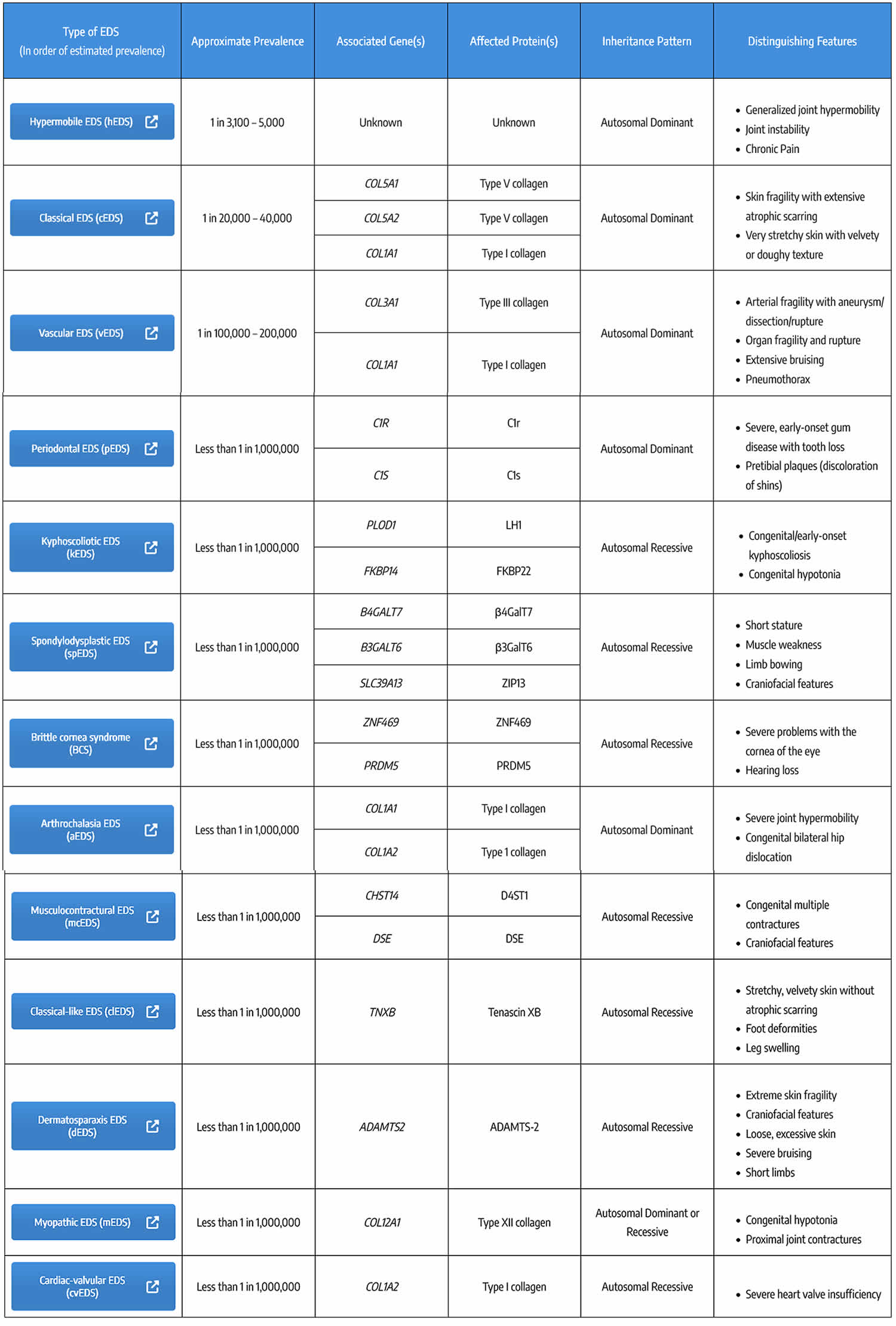
Footnotes: * Inheritance Pattern: AD = autosomal dominant; AR = autosomal recessive
[Source 126 ]Marfan syndrome
Marfan syndrome (MFS) also known as Marfan’s syndrome is a inherited genetic disorder that affects the connective tissue in many parts of your body 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138, 139, 140, 141, 142, 143. Connective tissue provides strength and flexibility to structures such as bones, ligaments, muscles, blood vessels, skin, lungs and heart valves. Marfan syndrome generally affects the limbs, but can also affect the spine, sternum, eyes, heart and blood vessels. The signs and symptoms of Marfan syndrome vary widely in severity, timing of onset, and rate of progression. Because connective tissue is found throughout your body, Marfan syndrome can affect many parts of your body such as your heart and blood vessels (cardiovascular), skeletal (bones, and joints) and eye (ocular) systems. The 2 main features of Marfan syndrome are vision problems caused by a dislocated lens in one or both eyes (ectopia lentis) and defects in the large blood vessel that distributes blood from the heart to the rest of the body called the aorta. The aorta can weaken and stretch, which may lead to a bulge in the blood vessel wall (aortic aneurysm). Stretching of the aorta may cause the aortic valve to leak, which can lead to a sudden tearing of the layers in the aorta wall (aortic dissection). Approximately 80% of patients will develop aortic complications. Aortic aneurysm and aortic dissection can be life threatening and is the cause of death in 30-45% of individuals with Marfan’s syndrome. Many people with Marfan syndrome have additional heart problems including a leak in the valve that connects two of the four chambers of the heart (mitral valve prolapse) or the valve that regulates blood flow from the heart into the aorta (aortic valve regurgitation). Leaks in these heart valves can cause shortness of breath, fatigue, and an irregular heartbeat felt as skipped or extra beats (palpitations).
Individuals with Marfan syndrome are usually tall and slender and have an arm span that exceeds their body height due to an overgrowth of the long bones of the arms and legs. Individuals with Marfan syndrome also have abnormally long slender fingers and toes resembling a spider’s legs (arachnodactyly), loose joints (joint hypermobility), long and narrow face, crowded teeth, abnormal side-to-side curvature of the spine (scoliosis) or forward curvature of the spine resulting in a “hunchback” or rounded appearance (kyphosis), stretch marks (striae) not related to weight gain or loss, and either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum). Some individuals develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax). A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia) in people with Marfan syndrome. Dural ectasia can cause pain in the back, abdomen, legs, or head. Most individuals with Marfan syndrome have some degree of nearsightedness (myopia). Clouding of the lens of the eye (cataract) may occur in mid-adulthood, and increased pressure within the eye (glaucoma) occurs more frequently in people with Marfan syndrome than in those without the condition. Marfan’s syndrome does not affect intelligence.
Note that these signs and symptoms and the severity of Marfan syndrome vary greatly from person to person. The features of Marfan syndrome can become apparent anytime between infancy and adulthood. Patients may be diagnosed with Marfan’s syndrome at birth, during childhood or later in life. The more severe cases are usually diagnosed early. Many patients present with features such as tall stature and an arm span that exceeds height. However, eye lens dislocation, aortic dilatation or skeletal signs and symptoms such as chest deformities (pectus excavatum or pectus carinatum) or curvature of the spine (scoliosis or kyphosis) may also be the first presentation. Many of the signs and symptoms of Marfan’s syndrome change with age so diagnosis can be difficult in children.
Depending on the onset and severity of signs and symptoms, Marfan syndrome can be fatal early in life; however, with proper treatment, many affected individuals have normal lifespans surviving into mid- to late adulthood.
If you have Marfan syndrome, it is important to understand how it affects your body and to be aware of your physical limitations. You can help prevent unnecessary stress or strain on joints by taking care during physical activity.
Marfan syndrome treatment usually includes medications to lower your blood pressure to reduce the strain on your aorta. Some people may need annual check-ups to monitor the heart. Regular monitoring to check for damage progression is vital. Many people with Marfan syndrome eventually require preventive surgery to repair damaged heart valves, blood vessels and joints.
Marfan syndrome treatment guidelines for all adults 128, 144:
- Restriction of physical activity with avoidance of contact sports, isometric exercise, and activities that can cause joint injury/pain
- Avoidance of agents that stimulate the cardiovascular system, such as decongestants and caffeine
- Annual ophthalmologic examination – LASIK correction of refractive errors is not recommended.
- Subacute endocarditis prophylaxis for dental work in the presence of mitral or aortic valve regurgitation
- Annual echocardiography to evaluate the ascending aorta in cases of small aortic dimensions and/or a slow aortic dilatation rate – Echocardiography is required with greater frequency when aortic root diameter is over 4.5 cm in adults, if aortic dilatation is greater than 0.5 cm per year, and if significant aortic regurgitation is present
- Beta-blockers or angiotensin receptor blockers (ARBs) treatment to reduce hemodynamic stress on the aortic wall. Current American College of Cardiology/American Heart Association guidelines recommend use of either beta blockers or angiotensin receptor blockers (ARBs) or a combination of both at maximally tolerated doses 145. European guidelines recommend use of beta blockers and consider angiotensin receptor blockers (ARBs) as an alternative treatment 146.
Marfan syndrome is caused by changes or mutations (variants) in the fibrillin-1 (FBN1) gene in the long arm of chromosome 15 at position 21.1 (15q21.1) 147.
About 3 out of 4 people (75% of patients) with Marfan syndrome inherit it, meaning they get the fibrillin-1 (FBN1) genetic mutation from a parent who has it. Most Marfan syndrome follows an autosomal dominant inheritance pattern, meaning that only one copy of a FBN1 gene variant is sufficient to cause Marfan syndrome. Autosomal dominant refers to a pattern of genetic inheritance where a genetic trait or condition is passed on when only one copy of a mutated gene (FBN1 gene) is inherited from a single parent. “Autosomal” means the gene is located on one of the numbered chromosomes (not sex chromosomes), and “dominant” means the altered gene is expressed even when a working copy is also present. People with an autosomal dominant condition have a 50% chance of passing the altered gene to each child. Meaning there is a 50 percent chance that a person with Marfan syndrome will pass along the genetic mutation each time they have a child. A child with a parent who has the mutated FBN1 gene has a 50% chance of inheriting Marfan syndrome. Both males and females are equally likely to inherit and be affected by autosomal dominant conditions. However, there are rare case reports describing autosomal recessive fibrillin 1 gene (FBN1) mutations 148. In autosomal recessive pattern of genetic inheritance, Marfan syndrome only appears only in individuals who have inherited 2 copies of a mutated or altered FBN1 gene, one from each parent, located on a non-sex (autosomal) chromosome. Both parents of an affected individual are typically “carriers,” possessing one mutated copy and one working copy of the FBN1 gene, but do not themselves show symptoms of Marfan syndrome. In autosomal recessive inheritance if a child inherits one mutated FBN1 gene and one working copy, the child will be a carrier but will not have Marfan syndrome.
About 25% of patients with Marfan syndrome are the first in their family to have it, when this happens it is called a spontaneous mutation or “de novo mutation” 149, 150. These spontaneous mutation or “de novo mutation” Marfan syndrome cases are more severely affected. A de novo mutation is a genetic alteration that appears for the first time in an individual and is not inherited from either parent. These mutations can occur in a parent’s egg or sperm cell, or in the fertilized egg itself during early development, leading to a new genetic change in that person. De novo mutations are responsible for some genetic disorders and diseases, as they represent fresh genetic changes that were not present in the prior generations of a family.
In less than 10% of patients with typical Marfan phenotype, no mutation in FBN1 is identifiable, likely due to complete allele deletion or altered regulation of the FBN1 gene 128. In patients with atypical presentations reminiscent of Marfan syndrome, a mutation in a gene encoding for transforming growth factor-beta receptor (TGFBR) may be the cause 128. Some individuals with TGFBR1 (Transforming Growth Factor Beta Receptor 1) or TGFBR1 (Transforming Growth Factor Beta Receptor 2) gene mutations have clinical features consistent with Marfan syndrome, while others have features of 1 of 2 other syndromes such as Loeys-Dietz syndrome (LDS) or familial thoracic aortic aneurysm (FTAA) syndrome 127.
The FBN1 gene provides instructions for making a protein called fibrillin-1 151. Fibrillin-1 attaches (binds) to other fibrillin-1 proteins and other molecules to form threadlike filaments called microfibrils 129. Microfibrils become part of the fibers that provide strength and flexibility to connective tissue 129. Additionally, microfibrils bind to molecules called growth factors and release them at various times to control the growth and repair of tissues and organs throughout the body 129. A mutation in the FBN1 gene can reduce the amount of functional fibrillin-1 that is available to form microfibrils, which leads to decreased microfibril formation 152, 153. As a result, microfibrils cannot bind to growth factors, so excess growth factors are available and elasticity in many tissues is decreased, leading to overgrowth and instability of tissues in Marfan syndrome 129.
Approximately 1 per 3,000 to 1 per 5,000 individuals is affected by Marfan syndrome 154 Marfan syndrome occurs worldwide, with no preference for race or gender. Marfan syndrome exhibits complete penetrance with variable expression 155.
Figure 16. Marfan syndrome arm span that exceeds their body height

Figure 17. Marfan syndrome – sunken chest (pectus excavatum)
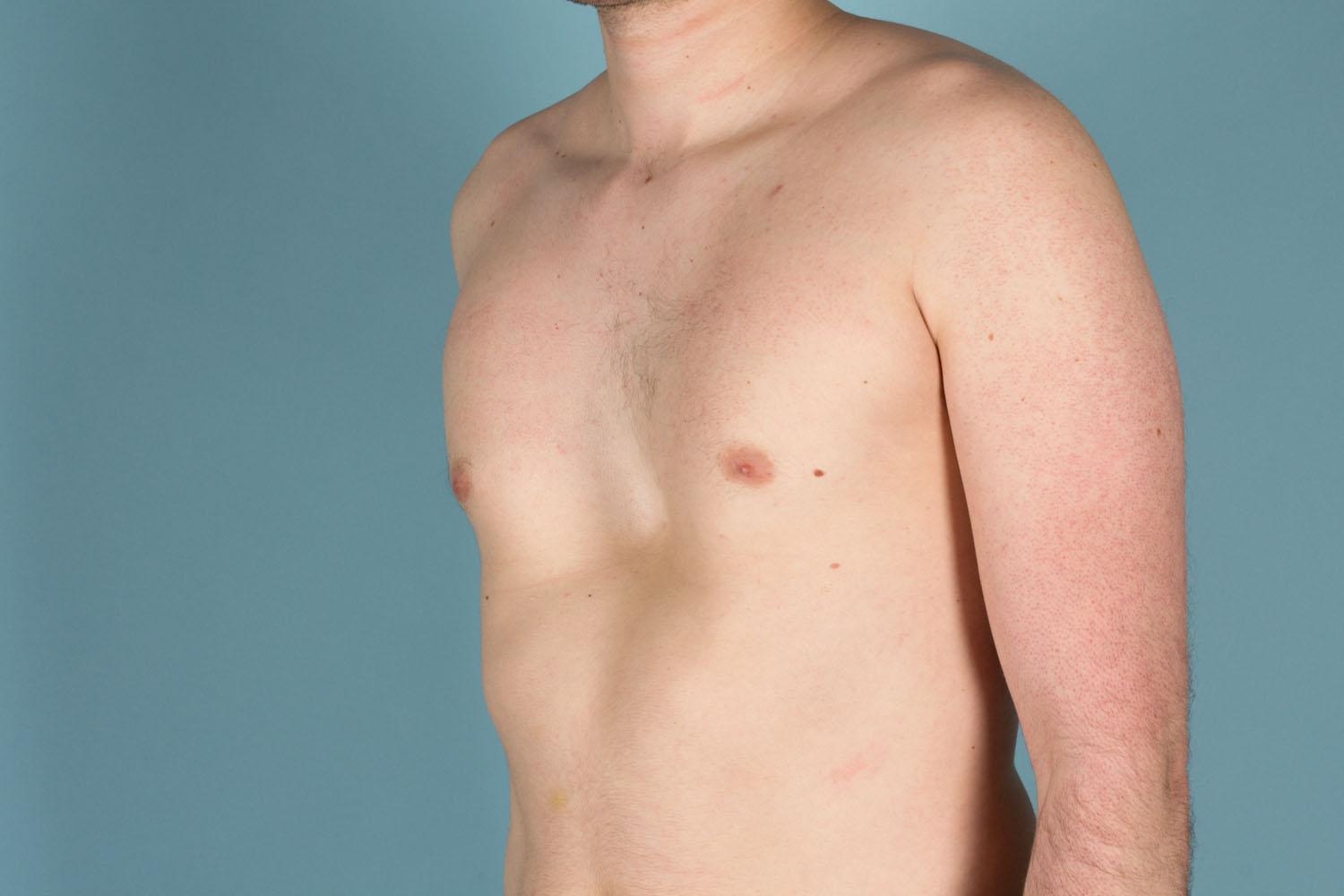
Figure 18. Marfan syndrome – protruding chest (pectus carinatum)
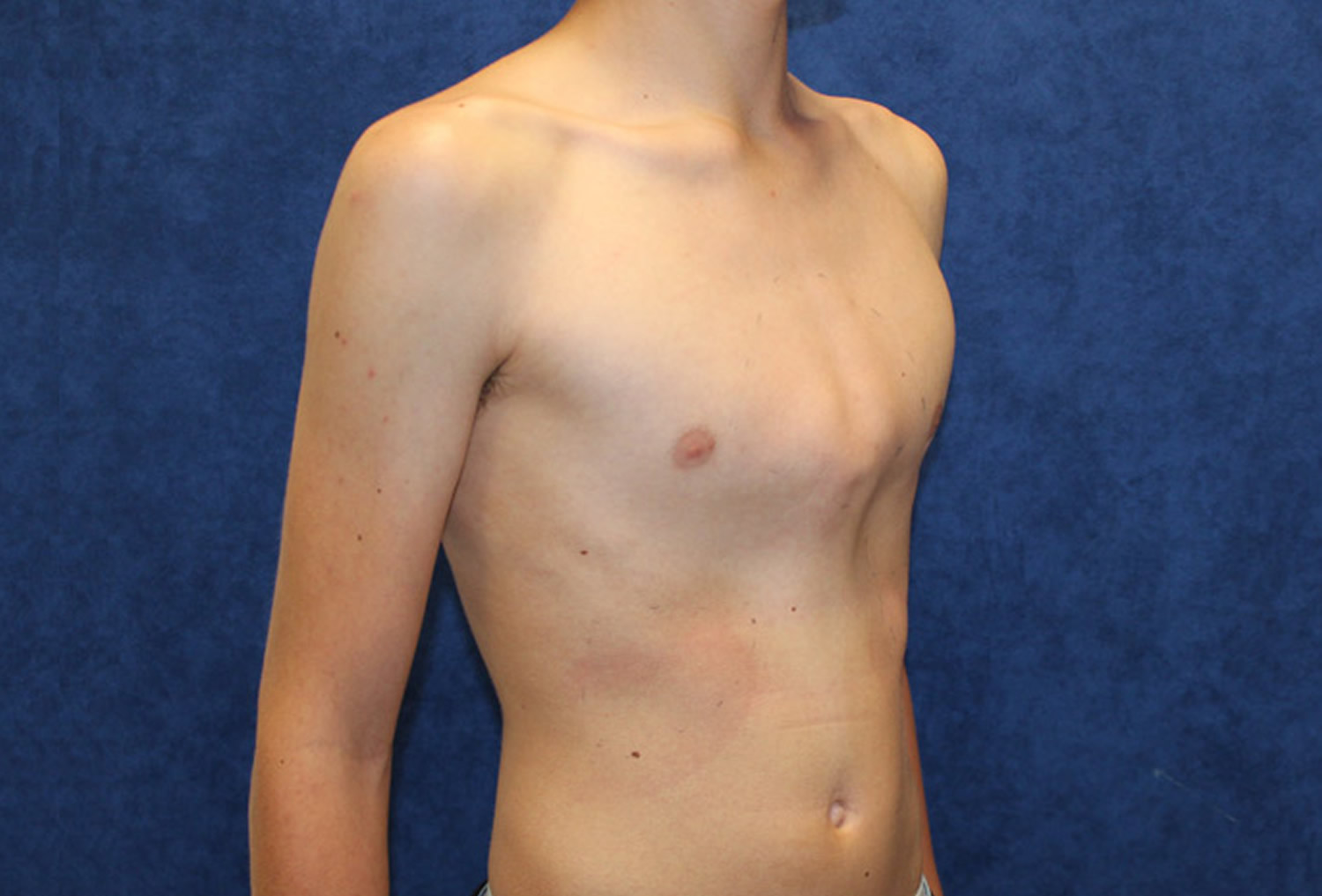
Figure 19. Arachnodactyly
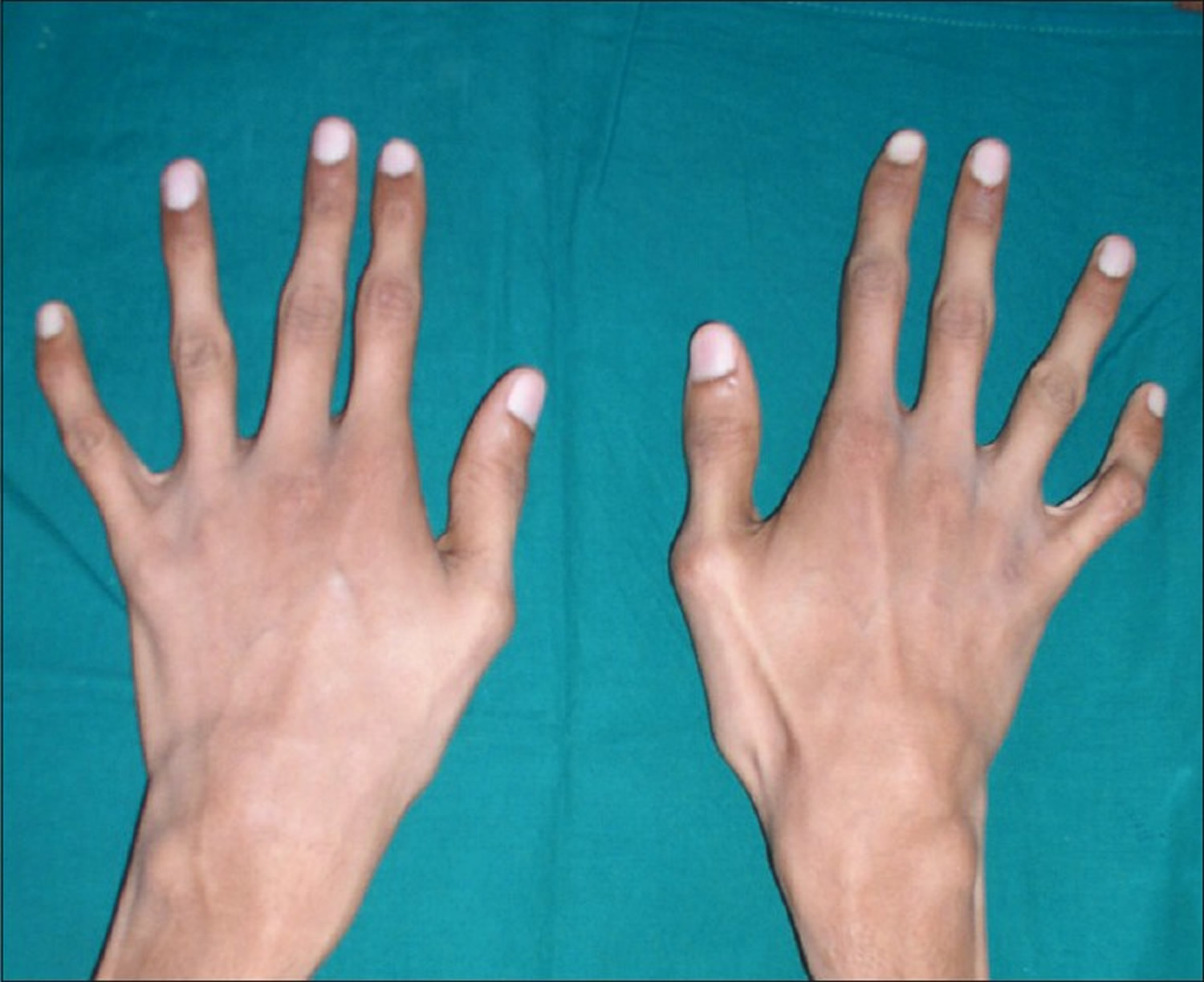
Loeys-Dietz syndrome
Loeys-Dietz syndrome (LDS) is a inherited connective tissue disorder that causes aortic aneurysms, widely spaced eyes (hypertelorism), a uvula (the little piece of flesh that hangs down in the back of the mouth) that splits in two (bifid uvula) and/or cleft palate and tortuous arteries (twisting or spiraled arteries) 156, 157, 158. Connective tissue is a catchall term for tissues composed of collagen and elastin that hold your body together and link its different parts. Connective tissue provides strength and flexibility to structures such as tendons, ligaments, skin, cartilage, bones, fascia and blood vessels, which provide support and structure to other tissues and organs. Connective tissue is essential to maintain the structure of your body. You have different forms of connective tissue nearly everywhere in your body. Other findings in people with Loeys-Dietz syndrome (LDS) include craniosynostosis (a condition in which one or more of the fibrous sutures in a young infant’s skull prematurely fuses or closes by turning into bone too early before the brain is fully formed thereby changing the growth pattern of the skull), exotropia (eyes that turn outward), micrognathia or mandibular hypoplasia (a condition in which the lower jaw or the mandible is smaller than usual), structural brain abnormalities, intellectual deficit, and congenital heart disease 156, 157, 158. Individuals with Loeys-Dietz syndrome are also predisposed to widespread and aggressive arterial aneurysms and pregnancy-related complications including uterine rupture and death 156. Individuals with Loeys-Dietz syndrome can show a strong predisposition for allergic/inflammatory disease including asthma, eczema, and reactions to food or environmental allergens 156. People with Loeys-Dietz syndrome (LDS) also have an increased incidence of gastrointestinal inflammation including eosinophilic esophagitis and eosinophilic gastritis and inflammatory bowel disease 156.
Loeys-Dietz syndrome (LDS) was was first observed and described by Dr. Bart Loeys and Dr. Hal Dietz at the Johns Hopkins University School of Medicine in 2005 and has many shared characteristics with Marfan syndrome 157. The prevalence of Loeys-Dietz syndrome is unknown but is estimated to be 1 per 50,000 newborns 158. More than 1,000 families with Loeys-Dietz syndrome have been described in the literature until 2018. Though with more availability of genetic testing the number of patients diagnosed has increased significantly in recent years. Loeys-Dietz syndrome occurs in all ethnic groups. Loeys-Dietz syndrome is a rare disorder that affects males and females in equal numbers.
Loeys-Dietz syndrome signs and symptoms vary among individuals. There are 7 types of Loeys-Dietz syndrome, labelled types 1 through 7, which are distinguished by their genetic cause. Loeys-Dietz syndrome is a genetic disorder that is caused by a mutation (gene change) in either the TGFBR1 or TGFBR2 genes (transforming growth factor beta receptor 1 or 2), the SMAD2 or SMAD3 genes (mothers against decapentaplegic homolog 2 or 3), the TGFB2 gene (tgfbeta 2 or transforming growth factor beta 2 ligand), the TGFB3 gene (tgfbeta 3 or transforming growth factor beta 3 ligand) or the IPO8 (Importin 8) gene 156.
- Loeys-Dietz syndrome 1 caused by mutations in the TGFBR1 gene
- Loeys-Dietz syndrome 2 caused by mutations in the TGFBR2 gene
- Loeys-Dietz syndrome 3 caused by mutations in the SMAD3 gene
- Loeys-Dietz syndrome 4 caused by mutations in the TGFB2 gene
- Loeys-Dietz syndrome 5 caused by mutations in the TGFB3 gene
- Loeys-Dietz syndrome 6 caused by mutations in the SMAD2 gene
- Loeys-Dietz syndrome 7 caused by mutations in the IPO8 gene
Although there is significant overlap between the clinical features caused by mutations in the different genes, scientists are learning about what might be differing features between the types and how this may impact medical management. Loeys-Dietz syndrome types 1 and 2 appear to be the most common forms. This condition is called Loeys-Dietz syndrome type 1 when affected individuals have cleft palate, craniosynostosis, and/or hypertelorism. Individuals without these features are said to have Loeys-Dietz syndrome type 2. Regardless of the type, signs and symptoms of Loeys-Dietz syndrome can become apparent anytime from childhood through adulthood, and the severity is variable.
These seven genes, TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, TGFB3 and bi-allelic variants in IPO8, play roles in a cell signaling pathway called the transforming growth factor beta (TGF-β) pathway, which directs the functions of the body’s cells during growth and development 159. This pathway also regulates the formation of the extracellular matrix, an intricate lattice of proteins and other molecules that forms in the spaces between cells and is important for tissue strength and repair. Mutations in the TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2 or TGFB3 gene result in the production of a protein with reduced function. Even though the protein is less active, signaling within the transforming growth factor beta (TGF-β) pathway occurs at an even greater intensity than normal in tissues throughout the body. Researchers speculate that the activity of other proteins in this signaling pathway is increased to compensate for the protein whose function is reduced; however, the exact mechanism responsible for the increase in signaling is unclear. The overactive transforming growth factor beta (TGF-β) pathway disrupts the development of the extracellular matrix and various body systems, leading to the signs and symptoms of Loeys-Dietz syndrome.
In 25 percent of Loeys-Dietz syndrome cases, Loeys-Dietz syndrome is inherited in an autosomal dominant manner with variable clinical expression and an affected person inherits the mutation from one affected parent 160. However, in about 75 percent of cases, Loeys-Dietz syndrome results from a new gene mutation and occurs in people with no history of the disorder in their family. This is called a de novo mutation.
It is important to have an early and adequate treatment for the heart problems because the chance for aortic dissection and other vascular problems may be high in some patients. Aortic dissection has been observed in early childhood (age ≥6 months) and/or at aortic dimensions that do not confer risk in other connective tissue disorders such as Marfan syndrome 160. Many specialists may be involved for the best management of the patient.
In Loeys-Dietz syndrome the aorta can weaken and stretch, causing a bulge in the blood vessel wall (an aneurysm). Stretching of the aorta may also lead to a sudden tearing of the layers in the aorta wall (aortic dissection). People with Loeys-Dietz syndrome can also have aneurysms or dissections in arteries throughout the body and have arteries with abnormal twists and turns (arterial tortuosity).
Individuals with Loeys-Dietz syndrome often have skeletal problems including premature fusion of the skull bones (craniosynostosis), an abnormal side-to-side curvature of the spine (scoliosis), either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum), an inward- and upward-turning foot (clubfoot), flat feet (pes planus), or elongated limbs with joint deformities called contractures that restrict the movement of certain joints. A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia). In individuals with Loeys-Dietz syndrome, dural ectasia typically does not cause health problems. Malformation or instability of the spinal bones (vertebrae) in the neck is a common feature of Loeys-Dietz syndrome and can lead to injuries to the spinal cord. Some affected individuals have joint inflammation (osteoarthritis) that commonly affects the knees and the joints of the hands, wrists, and spine.
People with Loeys-Dietz syndrome may bruise easily and develop abnormal scars after wound healing. The skin is frequently described as translucent, often with stretch marks (striae) and visible underlying veins. Some individuals with Loeys-Dietz syndrome develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax) or a protrusion of organs through gaps in muscles (hernias). Other characteristic features include widely spaced eyes (hypertelorism), eyes that do not point in the same direction (strabismus), a split in the soft flap of tissue that hangs from the back of the mouth (bifid uvula), and an opening in the roof of the mouth (cleft palate).
Individuals with Loeys-Dietz syndrome frequently develop immune system-related problems such as food allergies, asthma, or inflammatory disorders such as eczema or inflammatory bowel disease.
There is still no cure for Loeys-Dietz syndrome. Treatment focuses on managing the specific symptoms you have. These treatments typically require a team of specialists including a geneticist, cardiologist, heart (cardiothoracic) and bone (orthopedic) surgeons, ophthalmologist, rheumatologist, among others. It is recommended that you consult your physician or a local geneticist if you have questions about your health concerns. It is always important to consult your doctor to determine an effective and personalized course of action.
Most importantly, the goal of Loeys-Dietz syndrome treatment is to screen and correct blood vessel weaknesses before they tear. Your doctor may prescribe a beta-blocker or angiotensin receptor blocker (ARB) such as losartan to slow down the ballooning or stretching of the aortic root. Every 6 months to 1 year, you should be screened for aneurysms of the entire arterial tree from your head to your hips. This screening includes blood vessel imaging by CT angiography (CTA) or MR angiography (MRA) and heart imaging by echocardiography. If repeated imaging of the entire arterial tree does not show any changes or concern for aneurysms, it may be acceptable to image less frequently. Your doctor will be looking to see whether the aortic root balloons to larger than 4 cm (1.5 inches), at which point he/she may recommend surgery to replace the ballooning section of the aorta and sometimes the aortic valve of the heart. This surgery is typically safe and effective in fixing the problem.
To treat some of the musculoskeletal abnormalities associated with Loeys-Dietz syndrome, your care team may recommend nonsurgical management of the symptoms such as bracing or surgical correction of the abnormalities. These abnormalities include an abnormally curved spine (scoliosis), an indented or protruding chest, and issues with the bones of the neck. Doctors will recommend X-rays and CT scans to determine whether surgery should be performed. These surgeries can be complicated and require close attention for complications afterwards.
Staying active and conditioned is recommended for individuals with Loeys-Dietz syndrome. Athletic goals and limits should be discussed with the cardiologist and the care team to determine an individualized health plan.
Figure 20. Loeys-Dietz syndrome features
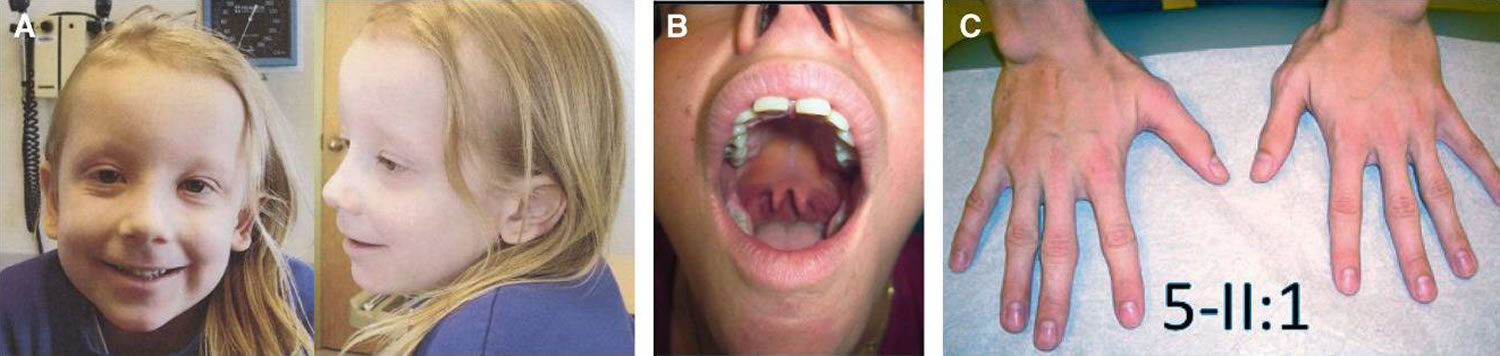
Footnotes: (A) hypertelorism, down-slanting palpebral fissures, amblyopia, and translucent skin in a 5-year-old girl, (B) bifid uvula in a 31-year-old woman, and (C) arachnodactyly in a 24-year-old man.
[Source 161 ]Stickler Syndrome
Stickler syndrome is a group of hereditary multisystem connective tissue disorder characterized by a distinctive facial appearance (mid-facial flatness, small chin, long upper lip or philtrum), palatal abnormalities (cleft palate, bifid uvula or high arched palate), eye abnormalities (vitreoretinal degeneration, myopia, cataracts, retinal holes and retinal detachments), hearing loss (sensorineural hearing loss), skeleton and joint problems (loose joints, scoliosis, chest deformities, a hip disorder of childhood [Legg-Calve-Perthe’s disease], early onset degenerative osteoarthritis before age 40 years) and mitral valve prolapse 162, 163, 164, 165, 166, 167, 168, 169, 170. These signs and symptoms present in Stickler syndrome often vary greatly from one individual to another. Stickler syndrome signs and symptoms can include eye findings of myopia, cataract, and retinal detachment (50% life-time risk of retinal detachment with minor eye trauma causing vitreous hemorrhage and/or retinal detachments); hearing loss that is both conductive and sensorineural; midfacial underdevelopment and cleft palate (either alone or as part of the Robin sequence); and mild spondyloepiphyseal dysplasia and/or precocious arthritis.
Stickler syndrome received its name from Dr. Gunnar B. Stickler, who first studied and documented the syndrome at Mayo Clinic in 1965 167. A syndrome is a collection of specific symptoms, all with one cause. Dr. Stickler called the disorder hereditary progressive arthro-ophthalmopathy. Connective tissue is the material between cells of the body that gives the tissue form and strength and is found all over the body. Connective tissue is made up of a protein known as collagen of which there are several different varieties found in the body. Stickler syndrome often affects the connective tissue of the eye, especially in the interior of the eyeball (vitreous humor), the specialized tissue that serves as a buffer or cushion for bones at joints (cartilage) and the ends of the bones that make up the joints of the body (epiphysis).
A characteristic feature of Stickler syndrome is a somewhat flattened facial appearance. This appearance results from underdeveloped bones in the middle of the face, including the cheekbones and the bridge of the nose. A particular group of physical features called Pierre Robin sequence is also common in people with Stickler syndrome. Pierre Robin sequence includes an opening in the roof of the mouth (a cleft palate), a tongue that is placed further back than normal (glossoptosis), and a small lower jaw (micrognathia). This combination of features can lead to feeding problems and difficulty breathing. Approximately 30% to 40% of patients with Pierre Robin sequence have Stickler syndrome 171, 172.
Many people with Stickler syndrome have severe nearsightedness (high myopia). In some cases, the clear gel that fills the eyeball (the vitreous) has an abnormal appearance, which is noticeable during an eye examination. Other eye problems are also common, including increased pressure within the eye (glaucoma), clouding of the lens of the eyes (cataracts), and tearing of the lining of the eye (retinal detachment). These eye abnormalities cause impaired vision or blindness in some cases.
In people with Stickler syndrome, hearing loss varies in degree and may become more severe over time. The hearing loss may be sensorineural, meaning that it results from changes in the inner ear, or conductive, meaning that it is caused by abnormalities of the middle ear.
Most people with Stickler syndrome have skeletal abnormalities that affect the joints. The joints of affected children and young adults may be loose and very flexible (hypermobile), though joints become less flexible with age. Arthritis often appears early in life and may cause joint pain or stiffness. Problems with the bones of the spine (vertebrae) can also occur, including abnormal curvature of the spine (scoliosis or kyphosis) and flattened vertebrae (platyspondyly). These spinal abnormalities may cause back pain.
Researchers have described several types of Stickler syndrome, which are distinguished by their genetic causes and their patterns of signs and symptoms. In particular, the eye abnormalities and severity of hearing loss differ among the types. Type I has the highest risk of retinal detachment. Type II also includes eye abnormalities, but type III does not (and is often called non-ocular Stickler syndrome). Types II and III are more likely than type I to have significant hearing loss. Types IV, V, and VI are very rare and have each been diagnosed in only a few individuals. Stickler syndrome affects an estimated 1 in 7,500 to 9,000 newborns 168, 169.
Table 2. Stickler syndrome types
| Stickler syndrome | Gene | Cytogenetic location | Distinguishing features |
|---|---|---|---|
| Type 1 | COL2A1 | 12q13.11 | Type 1. Membranous congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, deafness, arthropathy, cleft palate High risk of blindness |
| Ocular only | COL2A1 | 12q13.11 | Type 1. Membranous congenital vitreous anomaly, retinal detachment congenital megalophthalmos. No systemic features. High risk of blindness |
| Type 2 | COL11A1 | 1p21.1 | Beaded type 2 congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, deafness, arthropathy, cleft palate |
| Type 2 Recessive | COL11A1 | 1p21.1 | Autosomal recessive, Beaded congenital vitreous anomaly, retinal detachment, congenital megalophthalmos, cleft palate, profound severe congenital deafness |
| Type 3 | COL11A2 | 6p21.32 | Non-ocular Stickler |
| Type 4 | COL9A1 | 6q13 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 5 | COL9A2 | 1p34.2 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 6 | COL9A3 | 20q13.33 | Recessive inheritance, sensorineural deafness, myopia, vitreoretinopathy, retinal detachment, epiphyseal dysplasia |
| Type 7 | BMP4 | Hypoplastic vitreous, retinal detachment deafness, arthropathy, palate abnormality, renal dysplasia | |
| Type 8 | LOXL3 | 2p13.1 | Recessive inheritance Congenital myopia, hypoplastic vitreous, palate abnormality, Arthropathy Normal facies Normal hearing |
Table 3. Stickler Syndrome associated genes and its associated physical features
| Physical Feature | % of Persons with Physical Feature 1 | |||
|---|---|---|---|---|
| COL2A1 Stickler syndrome | COL11A1 Stickler syndrome | COL11A2 Stickler syndrome | COL9A1-, COL9A2-, & COL9A3 Stickler syndrome | |
| Cleft palate | 30%-60% | 60% | 35% | — |
| Myopia | 80%-90% | 80%-85% | — | 90%-95% |
| Retinal detachment | 40%-70% | <40% | — | 13%-18% |
| Hearing loss | 20%-50% (sensorineural hearing loss ± conductive hearing loss) | 75%-80% | 60% | 90%-95% (sensorineural hearing loss) |
| Skeletal signs and symptoms 2 | 35%-40% | 25% | 50% | 25% |
Footnotes: — = not reported; HL = hearing loss; SNHL = sensorineural hearing loss; SS = Stickler syndrome
2 Includes mainly early-onset degenerative joint disease
[Source 176 ]Stickler syndrome type I (STL1) is responsible for approximately 80% to 90% of reported Stickler syndrome cases (most common form of Stickler syndrome) and presents with a wide variety of symptoms affecting the eye, ear, facial appearance, palate and musculoskeletal system 177, 174. Most will have ‘full’ Stickler syndrome affecting the sight, joints, hearing and any mid-line clefting. Findings show those with this anomaly have an increased incidence of cleft abnormalities. Stickler syndrome type I (STL1) is caused to mutations over the entire COL2A1 gene on chromosome 12q13.11 178, 179, 180. These mutations cause loss of function of the COL2A1 gene. The majority of these mutations are associated with normal stature and early onset osteoarthritis. Only a few non-glycine missense mutations have been reported and among these, the arginine to cysteine substitutions predominate and these mutations cause some unusual disorders which may be described as Stickler-like but have short stature and brachydactyly. Stickler syndrome type I (STL1) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL2A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type I.
Stickler syndrome type II (STL2) occurs due to mutations of the COL11A1 gene on chromosome 1p21 178, 179, 180. Patients with another condition called Marshall syndrome can have mutations of COL11A1 gene also, but patients with Stickler syndrome type II have a milder physical feature with less prominent facial dysmorphism than patients with Marshall syndrome. Patients with Stickler syndrome type II have less pronounced midfacial flattening and the nasal bridge better developed than seen in patients with Marshall syndrome. Myopia and retinal degeneration are not always present. Cataracts and more severe early onset hearing loss are more common in Stickler type II than in patients with Stickler type I. Stickler syndrome type II (STL2) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A1 gene that is inherited from one parent is enough to cause the genetic condition. Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type II. There is also a autosomal recessive variety of Stickler syndrome type II (STL2) which has been identified in 3 people with very severe deafness.
Stickler syndrome type III (STL3) has been described as the non-eye form of Stickler syndrome, affecting the joints and hearing without involving the eyes. Stickler syndrome type III (STL3) is caused by mutations of the COL11A2 gene on chromosome 6p21.3 178, 179, 180. Stickler syndrome type III (STL3) is inherited in autosomal dominant pattern, meaning only one copy of a mutated (changed) COL11A2 gene that is inherited from one parent is enough to cause Stickler syndrome type III (STL3). Each child of an affected parent has a 50% chance of inheriting the mutated gene and Stickler syndrome type III (STL3). However, Stickler syndrome type III (STL3) is now considered the same disorder as heterozygous oto-spondylo-megaepiphyseal dysplasia (OSMED).
A mutation in a fourth gene, COL9A1, located on chromosome 6q13, has been identified in three reported intermarried families in Turkey and Morocco with Stickler syndrome type IV (STL4). The inheritance pattern is autosomal recessive meaning for a child to inherit Stickler syndrome type IV (STL4), the child need to receive two copies of a mutated COL9A1 gene, one from each parent. If a person only inherits one copy of the mutated COL9A1 gene, they are a carrier and usually do not show symptoms of Stickler syndrome type IV (STL4).
Stickler syndrome type V (STL5) is thought to be caused by COL9A2, located on chromosome 1p33. This has been described in one intermarried family in India. The inheritance pattern is autosomal recessive.
Mutations of COL9A3 have recently been reported in three brothers in an intermarried Moroccan family with features of Stickler syndrome and intellectual disability.
Stickler syndrome has also been subdivided based on the vitreous phenotype resulting from mutations in the various loci. However, it has been reported that it is difficult for most ophthalmologists to classify the type of vitreous anomalies in the patients with Stickler syndrome.
Stickler syndrome signs and symptoms
Eyes
- Short-sight or nearsightedness (myopia)
- Abnormal appearance of the vitreous gel.
- High risk of retinal detachments (tearing of the lining of the eye), which may affect both eyes.
- Cataracts
- Glaucoma
One of the first signs in Stickler syndrome is nearsightedness (myopia), in which objects close by are seen clearly but objects that are far away appear blurry. Myopia may vary from mild to severe in Stickler syndrome, but generally is not progressive and does not get worse 181. Myopia may be detectable shortly after birth, but the onset varies and may not develop until adolescence or even adulthood in some cases 181.
Bones and Joints
- Hyper-mobile (over flexible) joints and/or stiff joints.
- Early joint disease leading to osteoarthritis and joint replacements at a younger age
Facial Features
- A full cleft, submucous or high arched palate and/or bifid uvula
- Micrognathia – where the lower jaw is shorter than the other resulting in poor contact between the chewing surfaces of the upper and lower teeth. These symptoms are similar to those found in Pierre Robin sequence. It is reported that 30-40% of children diagnosed with Pierre Robin sequence are later re-diagnosed as having Stickler syndrome.
- Other facial characteristics include a flat face with a small nose and little or no nasal bridge. Appearance tends to improve with age
Hearing
- Hearing loss ( sensorineural and or conductive. The degree varies in affected individuals and may become more severe over time.
- Glue ear in childhood caused by cleft palate.
Other symptoms
These may include curvature of the spine (scoliosis), and because of sight and hearing problems, some learning difficulties may be experienced. Many people within the support group, especially children, complain of chronic fatigue.
A condition similar to Stickler syndrome, called Marshall syndrome, is characterized by a distinctive facial appearance, eye abnormalities, hearing loss, and early-onset arthritis. Marshall syndrome can also include short stature. Some researchers have classified Marshall syndrome as a variant of Stickler syndrome, while others consider it to be a separate disorder.
No studies to determine the prevalence of Stickler syndrome have been undertaken 163. However, an approximate incidence of Stickler syndrome among newborns can be estimated from data regarding the incidence of Robin sequence in newborns (1 per 10,000 to 1 per 14,000 newborns) and the percentage of these newborns who subsequently develop signs or symptoms of Stickler syndrome (35%). These data suggest that the incidence of Stickler syndrome among neonates is approximately 1 per 7,500 to 1 per 9,000 newborns 182. Stickler syndrome affects males as well as females.
Stickler is believed to be the most common connective tissue syndrome in the United States and Europe, but one of the rarest to be diagnosed. Most sufferers have such minor symptoms that they do not seek a diagnoses. Those who become patients are generally not correctly diagnosed. One study found a 53% error in original diagnosis of patients found in retrospect to have Stickler. A lot of patients are only diagnosed with one symptom and called, for example, arthritic or near-sighted.
The treatment of Stickler syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists including: geneticist, pediatrician and/or internist, orthopedic surgeon, rheumatologist, ophthalmologist and retina specialist, otolaryngologist, audiologist, plastic surgeon, orthodontist and other healthcare professionals may need to systematically and comprehensively plan an affected patient’s treatment.
Early identification of Stickler syndrome is important because it allows for surveillance and prompt treatment of associated abnormalities such as retinal detachment or skeletal malformations. Patients with ocular forms of Stickler syndrome are restricted from contact sports due to the risk of retinal detachment 162. Retinal detachment requires prompt surgery to preserve vision. Retinal detachment can recur even after successful surgery. Some physicians recommend prophylactic cryotherapy in certain cases to reduce the risk of developing retinal detachment.
Affected individuals should be made aware of the symptoms of retinal detachment so they can immediately have their eyes evaluated (ophthalmologic assessment) and treated if necessary. Surgery may also be necessary to remove cataracts.
Corrective lenses (glasses or contact lenses) are used to treat myopia.
Individuals with Stickler syndrome and Pierre-Robin sequence may require a tracheostomy (a procedure in which a tube is placed through a surgical opening in the neck) to prevent breathing (respiratory) difficulties. Surgery may also be required to fix various craniofacial abnormalities (e.g., cleft palate. micrognathia) that can contribute to breathing difficulties.
Orthodonture may be necessary to correct dental malalignment.
Patients with sensorineural or mixed hearing loss may require hearing aids. Hearing aids may be of benefit for certain individuals. A myringotomy, a surgical procedure in which a small incision is made in the eardrum and small tubes are inserted, may be used to treat glue ear. Various anti-inflammatory medications and sometimes prescription pain medications may be used to treat joint disease in individuals with Stickler syndrome. In mild cases, short-term relief may be provided from cortisone injections. Surgical correction of joint abnormalities may be necessary including joint replacement surgery such as a total hip or knee replacement. Surgery may also be necessary for skeletal malformations including abnormal curvature of the spine.
Physical therapy may prove beneficial in some cases.
Special education and other services may be helpful for children with learning disabilities due to hearing or vision problems.
Genetic counseling may be of benefit for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 21. Stickler syndrome
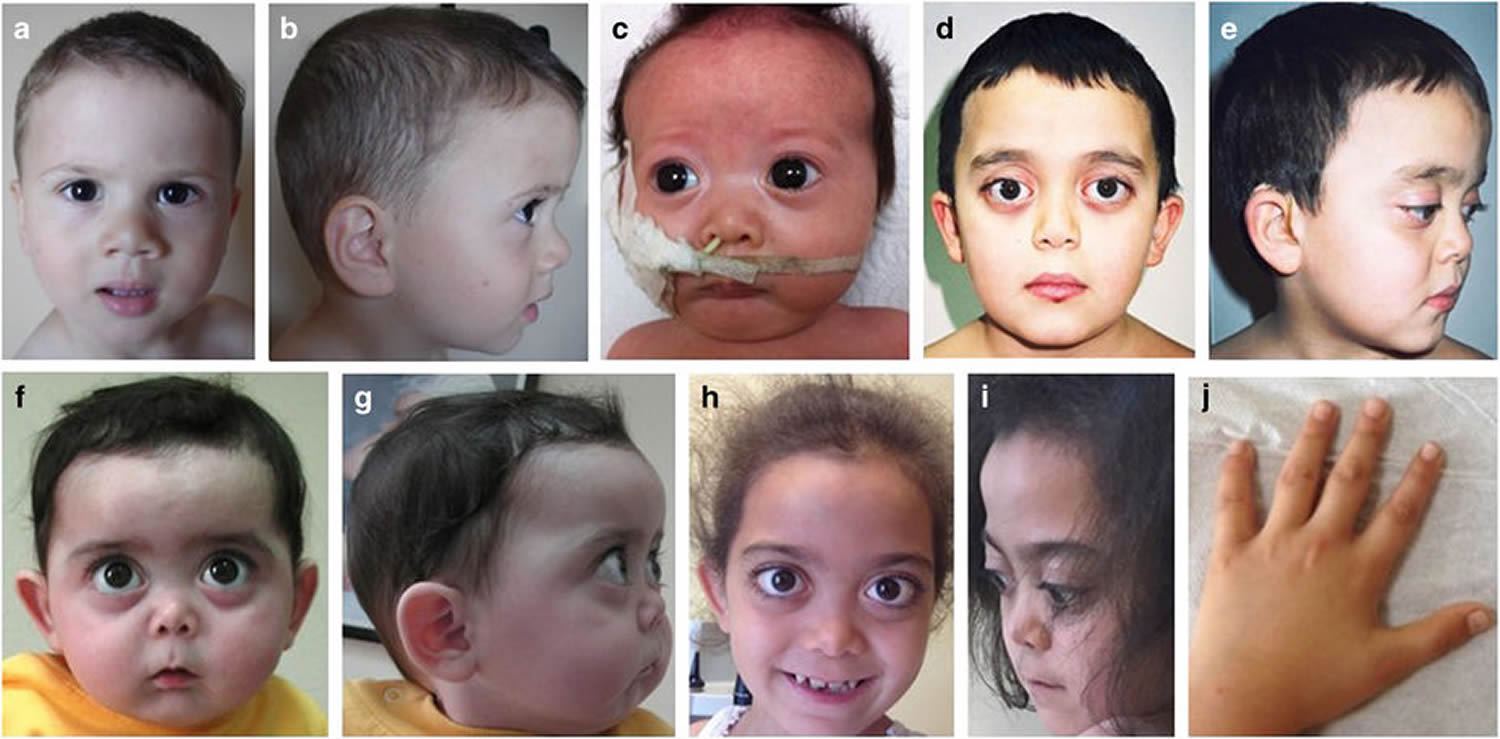
Note: Physical appearance of the patients. (a, b) Case 1, age 1.5 years. Mild depression of the nasal bridge and micrognathia. (c) Case 2, age 1 month. Buphthalmic eyes, hypertelorism, bilateral epicanthus, flat face, depressed nasal bridge, short stubby nose, and micro-retrognathia. (d, e) Case 4, age 8.5 years. Proptotic eyes, flat face with mild frontal bossing, depressed nasal bridge, and short nose. (f, g) Case 3, age 9 months. Buphthalmic eyes, flat face with frontal bossing, midfacial hypoplasia, depressed nasal bridge, short nose with anteverted nares, long philtrum, and micro-retrognathia. (h–j) Case 3, age 9 years. (h, i) High-frontal area, big proptotic eyes, long palpebral fissures, depressed nasal bridge, short nose, long philtrum, irregular teeth order, micrognathia, and dry rough hairs. (j) Small hands with brachydactyly.
Figure 22. Stickler syndrome joint hypermobility
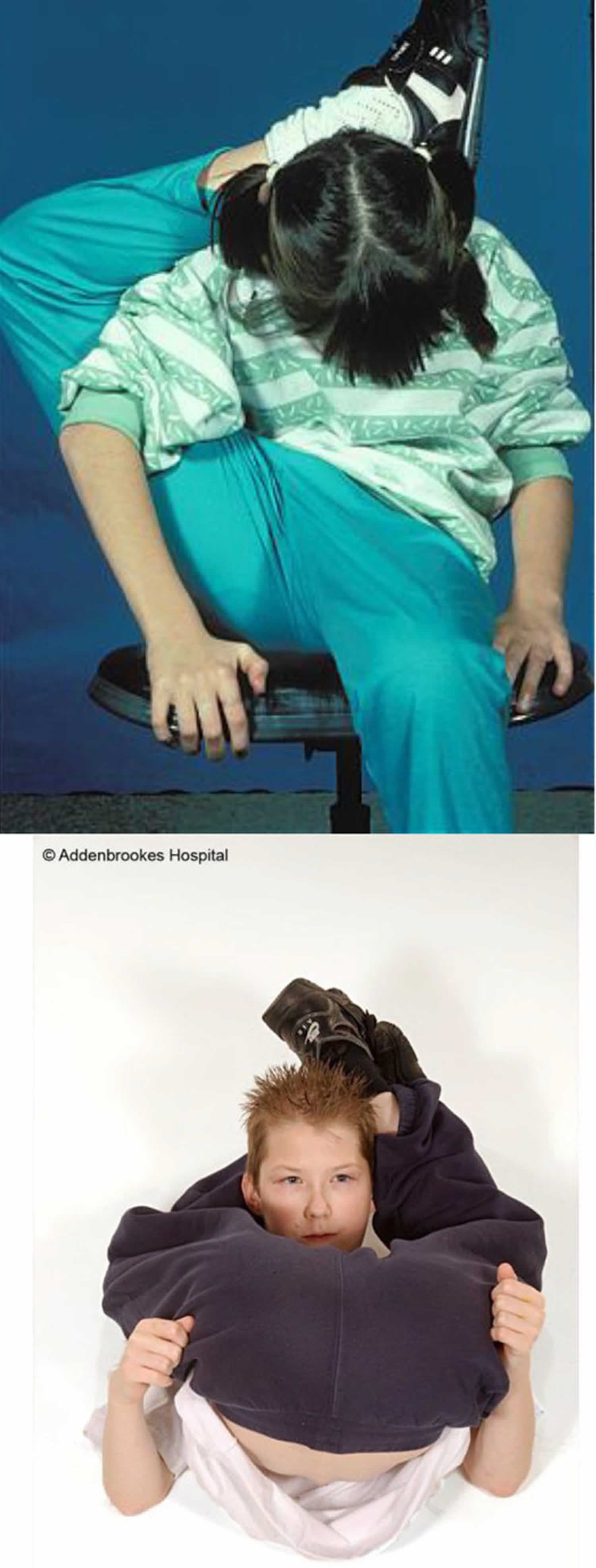
Figure 23. Stickler syndrome eye features
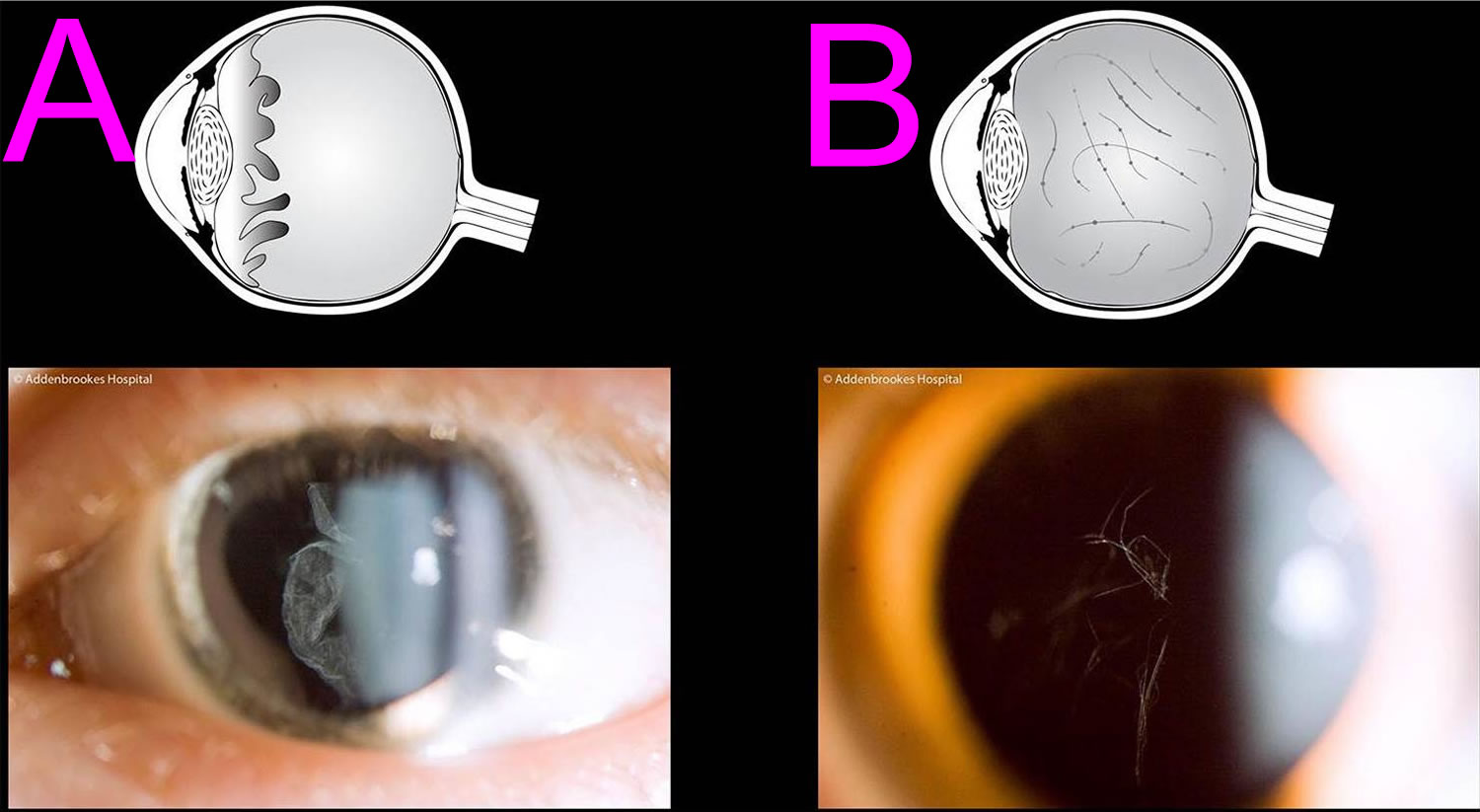
Footnotes: The pathognomonic hallmark of all but one of the sub-groups of Stickler syndrome (the only exception being Stickler syndrome type 3 where there is no eye involvement) is a congenital abnormality of vitreous embryogenesis. The secondary vitreous is normally fully matured by 10–14 weeks of intra-uterine growth and the embryogenic abnormalities in Stickler syndrome provide the clinician with a crucial sign on which to make the diagnosis. Stickler syndrome vitreoretinal abnormalities vary with the type of Stickler syndrome, sometimes described as membranous, beaded, or hypoplastic. Stickler syndrome Type 1 characteristically displays an optically empty-appearing central vitreous cavity with a membranous vitreous component within the vestigial gel in the retrolental space. Stickler syndrome Type 2 typically displays a beaded vitreous with irregular, thickened lamellae. Stickler syndrome Type 2 may less commonly have a hypoplastic vitreous which appears optically empty or with irregular lamellae architecture 183. (A) Membranous congenital vitreous anomaly (Stickler syndrome Type 1 haploinsufficiency mutations COL2A1 gene); (B) Beaded congenital vitreous anomaly (Stickler syndrome Type 2 COL11A1 dominant negative mutations).
[Source 173 ]Soft Tissue Sarcomas
Soft tissue sarcoma is a rare type of cancer that develops in the body’s connective tissues or soft tissues, such as fat, muscle, nerves, fibrous tissues, blood vessels, deep skin tissues and tendons. Soft tissue sarcoma typically presents as a painless lump or swelling, but symptoms can vary depending on the location. While it can affect people of all ages, it’s more common in older adults and slightly more likely in men.
A soft tissue sarcoma can develop anywhere in the body, but most commonly in the:
- Thigh
- Shoulder
- Arm
- Pelvis
- Abdomen.
Sarcomas can also be found in the trunk, head and neck area, internal organs, and the area in back of the abdominal (belly) cavity known as the retroperitoneum 184. About half of soft tissue sarcomas start in an arm or leg. Most people notice a lump that’s grown over time (weeks to months). The lump may or may not hurt.
When sarcomas grow in the back of the abdomen (the retroperitoneum), the symptoms often come from other problems the tumor is causing. For instance, they may cause blockage or bleeding of the stomach or bowels. They can press on nerves, blood vessels, or nearby organs. They can grow large enough for the tumor to be felt in the belly. Sometimes the tumors cause pain. About 4 of 10 sarcomas begin in the abdomen (belly).
In rare cases, sarcomas can start in the chest or in the head or neck.
If you have any of the these problems, see a doctor right away:
- A new lump or a lump that’s growing (anywhere on your body)
- Abdominal pain that’s getting worse
- Blood in your stool or vomit
- Black, tarry stools (when bleeding happens in the stomach or bowels, the blood can turn black as it’s digested, and it might make the stool very black and sticky)
These symptoms are more often caused by things other than sarcoma, but they still need to be checked out by a doctor.
Malignant (cancerous) tumours have the potential to spread to other parts of the body through the blood stream or lymph vessels and form another tumour at a new site. This new tumour is known as secondary cancer or metastasis.
According to the World Health Organization (WHO) classification, there are more than 100 different histologic subtypes of soft tissue sarcomas. They are named after the abnormal cells that make up the sarcoma.
Rhabdomyosarcoma is the most common type of soft tissue sarcoma. It forms from young cell types called rhabdomyoblasts that make up skeletal muscles – muscles which are connected to the skeleton to form part of the mechanical system which moves the limbs and other parts of the body. Rhabdomyosarcomas are most common in younger children under 10 years of age. However, they can develop in teenagers and adults.
The most common sarcome in adults is undifferentiated pleomorphic sarcoma (UPS), which is made up of many abnormal spindle-shaped cells. It is aggressive so it often returns or spreads after treatment.
Other types of soft tissue sarcoma include 184:
- Adult fibrosarcoma usually affects fibrous tissue in the legs, arms, or trunk. It’s most common in people between the ages of 20 and 60, but can occur in people of any age, even in infants.
- Alveolar soft-part sarcoma is a rare cancer that mostly affects young adults. These tumors most commonly start in the legs.
- Angiosarcoma can start in blood vessels (hemangiosarcomas) or in lymph vessels (lymphangiosarcomas). These tumors sometimes start in a part of the body that has been treated with radiation. Angiosarcomas are sometimes seen in the breast after radiation therapy and in limbs with lymphedema.
- Clear cell sarcoma is a rare cancer that often starts in tendons of the arms or legs. Under the microscope, it has some features of malignant melanoma, a type of cancer that starts in pigment-producing skin cells. How cancers with these features start in parts of the body other than the skin is not known.
- Desmoplastic small round cell tumor is a rare sarcoma of teens and young adults. It’s found most often in the abdomen (belly).
- Epithelioid sarcoma most often starts in tissues under the skin of the hands, forearms, feet, or lower legs. Teens and young adults are often affected.
- Ewing sarcoma a rare type of cancer that develops in bones or the soft tissue around bones, primarily affecting children and young adults. It is characterized by the formation of tumors, often in the legs, pelvis, or chest wall. While the exact cause is unknown, genetic mutations are believed to play a role in the development of Ewing sarcoma.
- Fibromyxoid sarcoma also called Evans’ tumor, low-grade is a slow-growing cancer that most often starts as a painless growth in the trunk or arms and legs (particularly the thigh). It is more common in young to middle aged adults.
- Gastrointestinal stromal tumor (GIST) is a type of sarcoma that starts in the digestive tract.
- Kaposi sarcoma is a type of sarcoma that starts in the cells lining lymph or blood vessels.
- Leiomyosarcoma is a type of cancer that starts in smooth muscle tissue. These tumors often start in the abdomen, but they can also start in other parts of the body, such as the arms or legs, or in the uterus.
- Liposarcomas are malignant tumors of fat tissue. They can start anywhere in the body, but they most often start in the thigh, behind the knee, and inside the back of the abdomen (belly). They occur mostly in adults between 50 and 65 years old.
- Malignant mesenchymoma is a rare type of sarcoma that shows features of fibrosarcoma and features of at least 2 other types of sarcoma.
- Malignant peripheral nerve sheath tumors include neurofibrosarcomas, malignant schwannomas, and neurogenic sarcomas. These sarcomas start in the cells that surround a nerve.
- Myxofibrosarcomas, low-grade are most often found in the arms and legs of elderly people. They are most common in or just under the skin, and there might be more than one tumor.
- Rhabdomyosarcoma is the most common type of soft tissue sarcoma in children.
- Synovial sarcoma is a malignant tumor of the tissue around joints. The most common locations are the hip, knee, ankle, and shoulder. This tumor is more common in children and young adults, but it can occur in older people.
- Undifferentiated pleomorphic sarcoma (UPS) was once called malignant fibrous histiocytoma. It’s most often found in the arms or legs. Less often, it can start inside at the back of the abdomen (the retroperitoneum). This sarcoma is most common in older adults. It mostly tends to grow into other tissues around the place it started, but it can spread to distant parts of the body.
Scientists don’t know exactly what causes most soft tissue sarcomas , but they have found some risk factors that can make a person more likely to develop these cancers. And research has shown that some of these risk factors affect the genes in cells in the soft tissues. However, researchers still don’t know why most soft tissue sarcomas develop in people who have no apparent risk factors.
Soft tissue sarcoma risk factors include:
- Radiation given to treat other cancers. Radiation exposure accounts for less than 5% of sarcomas. But patients might develop sarcomas from radiation given to treat other cancers, like breast cancer or lymphoma. The sarcoma often starts in the part of the body that was treated with radiation. The average time between the radiation treatments and the diagnosis of a sarcoma is about 10 years.
- Genetic factors. Some rare, inherited conditions can put people more at risk of soft tissue sarcoma. These conditions include von Recklinghausen disease also known as neurofibromatosis, Li–Fraumeni syndrome, Gardner syndrome, Werner syndrome, Gorlin syndrome, Tuberous sclerosis and retinoblastoma. Most people know if one of these very rare conditions runs in their family, and if so, that they may pass it to their children. If you do not know of this condition in your family, then it is very unlikely that it is present. A small number of people develop sarcoma due to genetic changes that happen during their lifetime, rather than inheriting a faulty gene.
- Neurofibromatosis also known as von Recklinghausen disease usually runs in families and causes many benign (not cancer) tumors that form in nerves under the skin and in other parts of the body (These are called neurofibromas.) It’s caused by a defect (mutation) in genes called NF1 and NF2. About 5% of people with neurofibromatosis will develop a sarcoma in a neurofibroma.
- Gardner syndrome is a disease caused by defects in the APC gene. This syndrome is a type of familial adenomatous polyposis (FAP), and people with it get many polyps in the colon (and intestines) and have a high risk of getting colon cancer . It also causes problems outside the colon, including desmoid tumors.
- Li-Fraumeni syndrome is caused by inherited defects in the TP53 gene. People affected by this syndrome have a high risk of cancer, such as breast cancer, brain tumors, leukemia , and sarcomas. Still, only 10 to 20 out of 100 people with Li-Fraumeni syndrome will develop a soft tissue sarcoma. People with Li-Fraumeni syndrome are sensitive to the cancer-causing effects of radiation. So if they have a cancer that’s treated with radiation, they have a very high chance of developing a new cancer in the part of the body that was treated.
- Retinoblastoma is an eye cancer in children that can be caused by defects in the RB1 gene. Children with this gene defect also have a higher risk of developing bone or soft tissue sarcomas, especially if the retinoblastoma was treated with radiation.
- Werner syndrome is caused by defects in the RECQL2 gene. Children with this syndrome have problems like those seen in the elderly. These include cataracts, skin changes, and clogged heart arteries (arteriosclerosis) which can lead to heart attacks. They also have an increased risk of cancer, including soft tissue sarcomas.
- Gorlin syndrome also called nevoid basal cell carcinoma syndrome (NBCCS). It’s caused by defects in the PTCH1 gene. People with this syndrome have a high risk of developing many basal cell skin cancers. They also have an increased risk of fibrosarcoma and rhabdomyosarcoma.
- Tuberous sclerosis can be caused by a defect in the TSC1 and/or TSC2 gene. People with this syndrome often have seizures and learning problems. They get benign (not cancer) tumors in many different organs. They also have kidney problems, often along with a kidney tumor called angiomyolipoma. People with tuberous sclerosis have an increased risk of rhabdomyosarcoma.
- Damaged lymph system. Lymph is a clear fluid containing immune system cells that’s carried throughout the body by a series of lymph vessels. These vessels connect lymph nodes – small bean-shaped collections of immune system cells. When lymph nodes have been removed or damaged by radiation therapy, lymph fluid can build up and cause swelling. This is called lymphedema. Lymphangiosarcoma a malignant (cancer) tumor that develops in lymph vessels is a very rare complication of chronic lymphedema.
- Chemicals. Exposure to vinyl chloride (a chemical used in making plastics) is a risk factor for developing sarcomas of the liver, but it hasn’t been proven to cause soft tissue sarcomas. Arsenic has also been linked to a type of liver sarcoma but not soft tissue sarcoma. Exposure to dioxin and to herbicides that contain phenoxyacetic acid at high doses (such as might occur in people who work on farms) may also be risk factors, but this isn’t known for certain. There’s no evidence that herbicides (weed killers) or insecticides, at levels encountered by the general public, cause sarcomas.
Treatment for a soft tissue sarcoma will depend on the type, location, and stage of the cancer, as well as your overall physical health. The only way to cure a soft tissue sarcoma is to remove it with surgery, so surgery is part of the treatment for all soft tissue sarcomas whenever possible.
Connective tissue disease causes
Connective tissue diseases is an umbrella term for a wide range of disorders that affect your body’s connective tissues, the fibers that are composed of collagen and elastin that support and anchor your organs and other structures in your body, such as tendons, ligaments, skin, cartilage, bones, fascia and blood vessels, which provide support and structure to other tissues and organs. Connective tissue diseases can be divided into 2 main types depending on its origin:
- Inherited genetic disorders caused by gene defects like Marfan syndrome (a inherited genetic disorder that affects connective tissue most commonly the heart, eyes, blood vessels and skeleton; people with Marfan syndrome are usually tall and thin with unusually long arms, legs, fingers and toes) and Ehlers-Danlos syndrome (a group of 13 inherited genetic connective tissue disorders including joint hypermobility, skin hyperextensibility, and tissue fragility)
- Autoimmune disorders also called autoimmune diseases caused by the body’s immune system which normally helps protect your body from infection and disease attacking its own tissues (autoimmune) such as Systemic Lupus Erythematosus (Lupus; an autoimmune disease that can affect many parts of the body, including the skin, joints, kidneys, and brain), Rheumatoid Arthritis (a chronic autoimmune inflammatory condition that primarily affects the joints, causing pain, swelling, and stiffness) and Scleroderma (an autoimmune disease that causes hardening and tightening of the skin and can also affect internal organs).
- Sarcomas are cancerous (malignant) tumors that arise from cells that make up the connective tissues. Sarcomas are rare tumors that account for approximately 1% of tumors in humans. Soft tissue sarcomas can arise from fat, muscle, nerve, tendons and blood and lymph vessel tissue. For some sarcomas the tissue of origin is uncertain such as pleiomorphic undifferentiated sarcoma. Synovial sarcoma is a misnomer as it does not arise from synovial tissue (as was originally thought). Sarcomas can occur almost anywhere in the body, but the most common areas are the arms and legs, the back of the abdomen (retroperitoneum) and head and neck. Sarcomas can affect adults or children. These tumors are diverse with significantly different signs and symptoms, progression and often different treatment regimens. According to the World Health Organization (WHO) classification, there are more than 100 different histologic subtypes of soft tissue sarcomas. The exact, underlying cause of these tumors is not fully understood. Most likely, complex genetic and environmental factors play a role in their development.
For the most part, scientists don’t know the underlying reasons why these malfunctions occur. But certain risk factors might play a part in making you more vulnerable to these diseases. Severe infections that overstress your immune system and exposure to certain toxic chemicals are possible risk factors.
Connective tissue disease signs and symptoms
Connective tissue diseases can affect many organs in your body including your heart, lungs, kidneys, joints and gastrointestinal tract, potentially leading to serious complications. Different connective tissue diseases can cause a wide variety of different symptoms. Connective tissue disease symptoms can vary greatly but may include joint pain and stiffness, skin rashes, fatigue, shortness of breath, organ-specific problems and Raynaud’s phenomenon also known as Raynaud’s disease that causes decreased blood flow to your fingers and toes in response to cold temperatures or stress.
Many connective tissue disorders can affect your lungs and cardiovascular system. Your lungs have a lot of connective tissues and rely heavily on them. Blood vessels run through most of your connective tissues, and because they’re made of similar stuff, inflammation spreads easily between them.
As a result, many connective tissue diseases can cause musculoskeletal symptoms together with cardiopulmonary symptoms, like shortness of breath and changes to your blood pressure or heartbeat. If your blood vessels become inflamed, they can swell and break, causing unexplained bleeding.
Sarcomas are cancers of the connective tissues often won’t cause symptoms until they grow large enough to compress an organ or vessel. But some can cause bone pain or joint pain where they start. Others may appear as a lump under your skin that may or may not be tender. Most sarcomas can spread, causing more widespread symptoms. The signs and symptoms of soft tissue sarcomas can vary greatly from one person to another. Specific findings depend on numerous factors including the specific subtype, the exact location of the tumor, the extent of the tumor into nearby tissue or organs, the specific organs involved and whether the disease has remained localized or spread to other areas of the body (metastasized). Often, soft tissue sarcomas will not be associated with any noticeable symptoms early in the course of the disease. At some point, affected individuals may notice a slow-growing, painless mass in the affected area. Tumors can become very large and cause pain or symptoms related to pressing against (compression of) nearby organs, nerves or structures in the body. Large tumors in the arms or legs can lead to a sensation of numbness, burning or tingling in the hands or feet (paresthesia). Tumors in the stomach or abdomen can cause abdominal pain or blood in the stool. Swelling (edema) can occur in areas where tumors push up against lymph vessels. Tumors near the skin can damage the skin, causing open sores (ulceration).
Sometimes, soft tissue sarcomas can cause nonspecific symptoms. These are symptoms that are common to many different disorders and conditions. These symptoms include nausea, vomiting, loss of appetite, unintended weight loss, unexplained fatigue and a general feeling of poor health (malaise).
Connective tissue disease diagnosis
Your doctor may order various tests depending on what type of connective tissue disorder is suspected. They’ll first ask for your medical history and a family history, and will do a physical examination. Further tests may include:
- Imaging tests, such as X-rays and magnetic resonance imaging (MRI) scans.
- Tests for markers of inflammation, such as C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR).
- Tests for antibodies, especially for autoimmune conditions.
- Tests for dry eyes or dry mouth.
- Blood and urine tests.
- A tissue biopsy.
- Genetic testing: Identifies specific genetic mutations responsible for some connective tissue disorders.
A connective tissue disorder can take a decade or more to be correctly diagnosed. For example, Ehlers-Danlos syndromes (EDS) diagnosis is often delayed (mean of 14 years and as long as 28 years) 185. Multiple factors may contribute to such delays, including the multisystem, complex, and phenotypically variable nature of the disorders, which leads to a broadly “positive review of systems” such that medical providers are skeptical about patients’ experience of the disorders 186, 187. Despite this, early diagnosis is critical to improved outcomes in quality of life, in part due to a reduced incidence of injury, and earlier detection of complications due to more comprehensive and timely surveillance being made available. Improvements to care coordination across the board are also noted with a diagnosis.
Connective tissue disease treatment
Most connective tissue diseases are lifelong (chronic) conditions and the clinical course of individuals with a connective tissue disease is highly variable 188. Connective tissue diseases can manifest in a multitude of physical and mental impairments in virtually any organ system can make the disorders difficult to recognize and manage. Management usually involves supportive care, treatment of associated secondary impairments, and preventive measures to mitigate or prevent problems that may occur or worsen over time. Doctors do what they can to reduce the severity and treat the symptoms and complications of the connective tissue disease individually. Autoimmune diseases and sarcomas (connective tissue cancers) can go into remission — periods without any signs or symptoms. But they can also recur (return).
Long-term management of connective tissue diseases must be individualized (person-centered at each and every step), coordinated, collaborative, comprehensive and most importantly multi-disciplinary. Proper and active care coordination among healthcare providers, patients, and their families is crucial to optimize outcomes and enhance the quality of life for individuals with these complex disorders.
Connective tissue diseases long-term management may include:
- Multidisciplinary Approach: Involves specialists in genetics, cardiology, orthopaedics, ophthalmology, and other fields to include; pain specialists, neurology, surgeons and immunology
- Regular Monitoring: Cardiovascular health, skeletal integrity, and ocular health are areas of high importance
- Supportive Therapies: Physical therapy, occupational therapy, and pain management to improve quality of life
- Medical and surgical interventions: Necessary for severe complications like aortic aneurysms or severe scoliosis and spinal instabilities
- Genetic Counselling: Provides patients and their families with information about the inheritance patterns, risks of recurrence in future pregnancies, and the implications of the disorder.
- Prenatal Testing: Provides options for families who wish to understand the genetic status of their offspring
- Education of appropriate lifestyle modifications
- Psychosocial assessment and supports.
Doctors treat autoimmune diseases with a combination of anti-inflammatory drugs (corticosteroids) and immune system-repressing drugs (immunosuppressants) to stop the inflammation. Fish oils contain the omega−3 fatty acids eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) that are known to reduce inflammation in the body and improve hypertriglyceridemia. Some studies have found that fish oil supplements may ease rheumatoid arthritis pain and stiffness. Side effects can include nausea, belching and a fishy taste in the mouth. Fish oil can get in the way of medicines you take. So check with your rheumatologist before trying it.
Scientists are researching genetic therapies that may one day be able to reduce the effects of genetic connective tissue disorders. For now, doctors can only treat the symptoms. In addition to medications, they recommend regular low-stress exercise or physical therapy to help manage musculoskeletal pain.
Treatment for a soft tissue sarcoma will depend on the type, location, and stage of the cancer, as well as your overall physical health. The only way to cure a soft tissue sarcoma is to remove it with surgery, so surgery is part of the treatment for all soft tissue sarcomas whenever possible. Additional treatments for sarcomas (connective tissue cancers) include chemotherapy, radiation therapy, targeted drug therapy (uses drugs to target specific genes and proteins that are involved in the growth and survival of cancer cells) and immunotherapy (use of medicines to help a person’s own immune system recognize and destroy cancer cells more effectively). Once your soft tissue sarcoma treatment has finished, you will have regular check-ups to confirm that the cancer hasn’t come back. For some people soft tissue sarcoma does come back after treatment, which is known as a recurrence. This is most likely to happen within the first five years after treatment. If the cancer does come back, treatment will depend on where the cancer has returned in your body and may include a mix of surgery, chemotherapy and radiation therapy. In some cases of advanced cancer, treatment will focus on managing any symptoms, such as pain, and improving your quality of life without trying to cure the disease. This is called palliative treatment and it can be provided in the home, in a hospital, in a palliative care unit or hospice.
Connective tissue disease prognosis
The wide range of connective tissue disorders have very different prognosis (outlooks) and possible complications. Your may recommend lifestyle changes to help optimize your overall health. Scrupulous cold avoidance may help to reduce the risk of flares of Raynaud phenomenon.
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Care Services; Committee on Selected Heritable Disorders of Connective Tissue and Disability; Wedge RA, Cartaxo T, Spicer CM, et al., editors. Selected Heritable Disorders of Connective Tissue and Disability. Washington (DC): National Academies Press (US); 2022 Jul 8. Available from: https://www.ncbi.nlm.nih.gov/books/NBK584971[↩][↩]
- Jarukitsopa S, Hoganson DD, Crowson CS, Sokumbi O, Davis MD, Michet CJ Jr, Matteson EL, Maradit Kremers H, Chowdhary VR. Epidemiology of systemic lupus erythematosus and cutaneous lupus erythematosus in a predominantly white population in the United States. Arthritis Care Res (Hoboken). 2015 May;67(6):817-28. doi: 10.1002/acr.22502[↩][↩]
- Margery-Muir AA, Bundell C, Nelson D, Groth DM, Wetherall JD. Gender balance in patients with systemic lupus erythematosus. Autoimmun Rev. 2017 Mar;16(3):258-268. doi: 10.1016/j.autrev.2017.01.007[↩]
- Feldman CH, Costenbader KH. Epidemiology and classification of systemic lupus erythematosus. Chapter 133. In: Hochberg MC, Gravallese EM, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors(s). Rheumatology. Seventh edition. Philadelphia PA, USA: Elsevier, 2019:1091-5.[↩]
- Frances Rees, Michael Doherty, Matthew J Grainge, Peter Lanyon, Weiya Zhang, The worldwide incidence and prevalence of systemic lupus erythematosus: a systematic review of epidemiological studies, Rheumatology, Volume 56, Issue 11, November 2017, Pages 1945–1961, https://doi.org/10.1093/rheumatology/kex260[↩][↩]
- Yacoub Wasef SZ. Gender differences in systemic lupus erythematosus. Gend Med. 2004 Aug;1(1):12-7. doi: 10.1016/s1550-8579(04)80006-8[↩]
- Masoumi M, Solaymani M, Abbasifard M, Houshmandfar S, Iravani P, Saeedi-Boroujeni A, Karami J. The genetic puzzle of rheumatoid arthritis: Causes, progression, and treatment. Biochem Biophys Rep. 2025 Jul 21;43:102148. doi: 10.1016/j.bbrep.2025.102148[↩][↩]
- Di Matteo A, Bathon JM, Emery P. Rheumatoid arthritis. Lancet. (2023) 402:2019–33. doi: 10.1016/S0140-6736(23)01525-8[↩]
- Yang P, Song Y, Li M. Biological mechanisms of pulmonary inflammation and its association with seropositive rheumatoid arthritis. Front Immunol. 2025 May 23;16:1530753. doi: 10.3389/fimmu.2025.1530753[↩]
- Rheumatoid Arthritis. https://www.niams.nih.gov/health-topics/rheumatoid-arthritis[↩][↩]
- Bax M., van Heemst J., Huizinga T.W., Toes R.E. Genetics of rheumatoid arthritis: what have we learned? Immunogenetics. 2011;63:459–466. doi: 10.1007/s00251-011-0528-6[↩][↩]
- Weyand C. New insights into the pathogenesis of rheumatoid arthritis. Rheumatology. 2000;39(suppl_1):3–8. doi: 10.1093/oxfordjournals.rheumatology.a031491[↩]
- Mena-Vazquez N, Perez AL, Manrique-Arija S, Romero BC, Gomez CC, Urena GI, et al. Analysis of clinical-analytical characteristics in patients with rheumatoid arthritis and interstitial lung disease: case-control study. Reumatol Clin (Engl Ed). (2021) 17:197–202. doi: 10.1016/j.reuma.2019.06.001[↩]
- Liu YCLF. High levels of antibodies to citrullinated alpha-enolase peptide-1 (CEP-1) identify erosions and interstitial lung disease (ILD) in a Chinese rheumatoid arthritis cohort. Clin Immunol: Off J Clin Immunol Soc. (2019) 200:10–5. doi: 10.1016/j.clim.2019.01.001[↩]
- Giles JT, Danoff SK, Sokolove J, Wagner CA, Winchester R, Pappas DA, et al. Association of fine specificity and repertoire expansion of anticitrullinated peptide antibodies with rheumatoid arthritis associated interstitial lung disease. Ann Rheum Dis. (2014) 73:1487–94. doi: 10.1136/annrheumdis-2012-203160[↩]
- Bullock J, Rizvi SAA, Saleh AM, Ahmed SS, Do DP, Ansari RA, Ahmed J. Rheumatoid Arthritis: A Brief Overview of the Treatment. Med Princ Pract. 2018;27(6):501-507. doi: 10.1159/000493390[↩]
- Gravallese E.M., Firestein G.S. Rheumatoid arthritis—common origins, divergent mechanisms. N. Engl. J. Med. 2023;388(6):529–542. doi: 10.1056/NEJMra2103726[↩][↩]
- Masoumi M., Bashiri H., Khorramdelazad H., Barzaman K., Hashemi N., Sereshki H.A., et al. Destructive roles of fibroblast-like synoviocytes in chronic inflammation and joint damage in rheumatoid arthritis. Inflammation. 2021;44:466–479. doi: 10.1007/s10753-020-01371-1[↩]
- Masoumi M., Mehrabzadeh M., Mahmoudzehi S., Mousavi M.J., Jamalzehi S., Sahebkar A., Karami J. Role of glucose metabolism in aggressive phenotype of fibroblast-like synoviocytes: latest evidence and therapeutic approaches in rheumatoid arthritis. Int. Immunopharmacol. 2020;89 doi: 10.1016/j.intimp.2020.107064[↩]
- Okada Y., Eyre S., Suzuki A., Kochi Y., Yamamoto K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019;78(4):446–453. doi: 10.1136/annrheumdis-2018-213678[↩]
- Zhao J., Guo S., Schrodi S.J., He D. Cuproptosis and cuproptosis–related genes in rheumatoid arthritis: implication, prospects, and perspectives. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.930278[↩]
- Scherer H.U., Häupl T., Burmester G.R. The etiology of rheumatoid arthritis. J. Autoimmun. 2020;110 doi: 10.1016/j.jaut.2019.102400[↩]
- Silman A.J., Pearson J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. Ther. 2002;4:1–8. doi: 10.1186/ar578[↩]
- Riaz M, Rasool G, Yousaf R, Fatima H, Munir N, Ejaz H. Anti-Rheumatic potential of biological DMARDS and protagonistic role of bio-markers in early detection and management of rheumatoid arthritis. Innate Immun. 2025 Jan-Dec;31:17534259251324820. doi: 10.1177/17534259251324820[↩][↩][↩][↩][↩][↩]
- InformedHealth.org [Internet]. Cologne, Germany: Institute for Quality and Efficiency in Health Care (IQWiG); 2006-. Overview: Rheumatoid arthritis. [Updated 2024 Jan 11]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK384455[↩][↩][↩][↩]
- Rheumatoid arthritis. https://www.mayoclinic.org/diseases-conditions/rheumatoid-arthritis/symptoms-causes/syc-20353648[↩]
- Rustamovich T.D., Alisherovna K.M., Djamshedovna K.D., Nizamitdinovich K.S. Features of the psychoemotional status of patients with rheumatoid arthritis. Miasto Przyszłości. 2023;32:23–30.[↩]
- Wu F, Gao J, Kang J, Wang X, Niu Q, Liu J, et al. B cells in rheumatoid arthritis:Pathogenic mechanisms and treatment prospects. Front Immunol. (2021) 12:750753. doi: 10.3389/fimmu.2021.750753[↩]
- Malmstrom V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. (2017) 17:60–75. doi: 10.1038/nri.2016.124[↩]
- Khudhair HAA. A study of the roles of some immunological biomarkers in the diagnosis of rheumatoid arthritis. J Med Life. 2023 Aug;16(8):1194-1200. doi: 10.25122/jml-2023-0158[↩][↩][↩][↩]
- Brown P, Pratt AG, Hyrich KL. Therapeutic advances in rheumatoid arthritis. BMJ. 2024 Jan 17;384:e070856. https://www.bmj.com/content/384/bmj-2022-070856.long[↩][↩][↩][↩][↩]
- Abedian Z, Sagafi M, Kenari SA, Abedian F. Anti-perinuclear factor as diagnostic marker in rheumatoid arthritis. J Clin Diagn Res. 2015;9(9):OC13–OC16. doi: 10.7860/JCDR/2015/14962.6456[↩]
- Kurki P, Aho K, Palosuo T, Heliövaara M. Immunopathology of rheumatoid arthritis. Antikeratin antibodies precede the clinical disease. Arthritis Rheum. 1992 Aug;35(8):914-7. doi: 10.1002/art.1780350810[↩]
- Vencovský J, Machácek S, Sedová L, Kafková J, Gatterová J, Pesáková V, Růzicková S. Autoantibodies can be prognostic markers of an erosive disease in early rheumatoid arthritis. Ann Rheum Dis. 2003 May;62(5):427-30. https://pmc.ncbi.nlm.nih.gov/articles/instance/1754544/pdf/v062p00427.pdf[↩]
- Saraux A, Berthelot JM, Devauchelle V, Bendaoud B, Chalès G, Le Henaff C, Thorel JB, Hoang S, Jousse S, Baron D, Le Goff P, Youinou P. Value of antibodies to citrulline-containing peptides for diagnosing early rheumatoid arthritis. J Rheumatol. 2003 Dec;30(12):2535-9. https://www.jrheum.org/content/jrheum/30/12/2535.full.pdf[↩]
- Vossenaar ER, Després N, Lapointe E, van der Heijden A, Lora M, Senshu T, van Venrooij WJ, Ménard HA. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res Ther. 2004;6(2):R142-50. doi: 10.1186/ar1149[↩]
- Renger F, Bang H, Feist E, Fredenhagen G, Natusch A, Backhaus M, Burmester GR, Egerer K. Immediate determination of ACPA and rheumatoid factor–a novel point of care test for detection of anti-MCV antibodies and rheumatoid factor using a lateral-flow immunoassay. Arthritis Res Ther. 2010;12(3):R120. doi: 10.1186/ar3057[↩]
- Innala L, Kokkonen H, Eriksson C, Jidell E, Berglin E, Dahlqvst SR. Antibodies against mutated citrullinated vimentin are a better predictor of disease activity at 24 months in early rheumatoid arthritis than antibodies against cyclic citrullinated peptides. J Rheumatol. 2008 Jun;35(6):1002-8. https://www.jrheum.org/content/jrheum/35/6/1002.full.pdf[↩]
- Luime J, Colin E, Hazes J, et al. Does anti-MCV has additional value as serological marker in the diagnostic and prognostic work-up of patients with rheumatoid arthritis? A systematic review. Ann Rheum Dis 2009. 10.1136/ard.2008.103283[↩]
- Wagner E, Skoumal M, Bayer PM, Klaushofer K. Antibody against mutated citrullinated vimentin: a new sensitive marker in the diagnosis of rheumatoid arthritis. Rheumatol Int. 2009 Sep;29(11):1315-21. doi: 10.1007/s00296-009-0854-2[↩]
- Corrigall VM, Bodman-Smith MD, Fife MS, Canas B, Myers LK, Wooley P, Soh C, Staines NA, Pappin DJ, Berlo SE, van Eden W, van Der Zee R, Lanchbury JS, Panayi GS. The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J Immunol. 2001 Feb 1;166(3):1492-8. doi: 10.4049/jimmunol.166.3.1492[↩]
- Bodman-Smith MD, Corrigall VM, Berglin E, Cornell HR, Tzioufas AG, Mavragani CP, Chan C, Rantapää-Dahlqvist S, Panayi GS. Antibody response to the human stress protein BiP in rheumatoid arthritis. Rheumatology (Oxford). 2004 Oct;43(10):1283-7. doi: 10.1093/rheumatology/keh312[↩]
- Zanwar A, Ahmed S, Misra DP, et al. Recent evidence comparing combination of conventional synthetic disease-modifying antirheumatic drugs with biologic disease-modifying antirheumatic drugs in rheumatoid arthritis. Indian J Rheumatol 2017; 12: 104–109.[↩]
- Adigun R, Goyal A, Hariz A. Systemic Sclerosis (Scleroderma) [Updated 2024 Apr 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430875[↩][↩]
- Sapkota B, Al Khalili Y. Mixed Connective Tissue Disease. [Updated 2024 Jul 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK542198[↩][↩][↩]
- Zandman-Goddard G, Solomon M, Rosman Z, Peeva E, Shoenfeld Y. Environment and lupus-related diseases. Lupus. 2012 Mar;21(3):241-50. doi: 10.1177/0961203311426568[↩]
- Sharp GC, Irvin WS, Tan EM, Gould RG, Holman HR. Mixed connective tissue disease–an apparently distinct rheumatic disease syndrome associated with a specific antibody to an extractable nuclear antigen (ENA). Am J Med. 1972 Feb;52(2):148-59. doi: 10.1016/0002-9343(72)90064-2[↩][↩]
- Bennett RM, O’Connell DJ. Mixed connective tisssue disease: a clinicopathologic study of 20 cases. Semin Arthritis Rheum. 1980 Aug;10(1):25-51. doi: 10.1016/0049-0172(80)90013-x[↩]
- Amigues JM, Cantagrel A, Abbal M, Mazieres B. Comparative study of 4 diagnosis criteria sets for mixed connective tissue disease in patients with anti-RNP antibodies. Autoimmunity Group of the Hospitals of Toulouse. J Rheumatol. 1996 Dec;23(12):2055-62.[↩]
- Alarcón-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol. 1989 Mar;16(3):328-34.[↩][↩]
- Alarcon-Segovia D, Villareal M. Classification and diagnostic criteria for mixed connective tissue disease. Kasukawa R, Sharp GC, eds. Mixed Connective Tissue Disease and Anti-Nuclear Antibodies. Amsterdam: Excerpta Medica; 1987. 33-40.[↩]
- Tanaka Y, Kuwana M, Fujii T, Kameda H, et al. 2019 Diagnostic criteria for mixed connective tissue disease (MCTD): From the Japan research committee of the ministry of health, labor, and welfare for systemic autoimmune diseases. Mod Rheumatol. 2021 Jan;31(1):29-33. https://doi.org/10.1080/14397595.2019.1709944[↩][↩]
- Kubo S, Tanaka Y. Evolution of diagnostic criteria and new insights into clinical testing in mixed connective tissue disease; anti-survival motor neuron complex antibody as a novel marker of severity of the disease. Immunol Med. 2024 Jun;47(2):52-57. doi: 10.1080/25785826.2024.2338593[↩]
- Gunnarsson R, Hetlevik SO, Lilleby V, Molberg Ø. Mixed connective tissue disease. Best Pract Res Clin Rheumatol. 2016 Feb;30(1):95-111. doi: 10.1016/j.berh.2016.03.002[↩]
- Cansu DÜ, Korkmaz C. Pulmonary hypertension in connective tissue diseases: epidemiology, pathogenesis, and treatment. Clin Rheumatol. 2023 Oct;42(10):2601-2610. doi: 10.1007/s10067-022-06446-y[↩]
- Cappelli S, Bellando Randone S, Martinović D, et al. “To be or not to be,” ten years after: evidence for mixed connective tissue disease as a distinct entity. Semin Arthritis Rheum. 2012 Feb;41(4):589-98. doi: 10.1016/j.semarthrit.2011.07.010[↩]
- Yoshida S. Pulmonary arterial hypertension in connective tissue diseases. Allergol Int. 2011 Dec;60(4):405-9. doi: 10.2332/allergolint.11-RAI-0360[↩]
- Gunnarsson R, El-Hage F, Aaløkken TM, Reiseter S, Lund MB, Garen T; Norwegian MCTD study group; Molberg Ø. Associations between anti-Ro52 antibodies and lung fibrosis in mixed connective tissue disease. Rheumatology (Oxford). 2016 Jan;55(1):103-8. doi: 10.1093/rheumatology/kev300[↩]
- Mixed Connective-Tissue Disease (MCTD). https://emedicine.medscape.com/article/335815-overview[↩][↩]
- Martínez-Barrio J, Valor L, López-Longo FJ. Facts and controversies in mixed connective tissue disease. Med Clin (Barc). 2018 Jan 12;150(1):26-32. English, Spanish. doi: 10.1016/j.medcli.2017.06.066[↩]
- Ciang NC, Pereira N, Isenberg DA. Mixed connective tissue disease-enigma variations? Rheumatology (Oxford). 2017 Mar 1;56(3):326-333. doi: 10.1093/rheumatology/kew265[↩]
- Alves MR, Isenberg DA. “Mixed connective tissue disease”: a condition in search of an identity. Clin Exp Med. 2020 May;20(2):159-166. doi: 10.1007/s10238-020-00606-7[↩]
- Maldonado ME, Perez M, Pignac-Kobinger J, Marx ET, Tozman EM, Greidinger EL, Hoffman RW. Clinical and immunologic manifestations of mixed connective tissue disease in a Miami population compared to a Midwestern US Caucasian population. J Rheumatol. 2008 Mar;35(3):429-37. https://pmc.ncbi.nlm.nih.gov/articles/PMC2919224[↩]
- Ungprasert P, Crowson CS, Chowdhary VR, Ernste FC, Moder KG, Matteson EL. Epidemiology of Mixed Connective Tissue Disease, 1985-2014: A Population-Based Study. Arthritis Care Res (Hoboken). 2016 Dec;68(12):1843-1848. doi: 10.1002/acr.22872[↩][↩]
- Nakae K, Furusawa F, Kasukawa R, et al. . A nationwide epidemiological survey on diffuse collagen diseases: Estimation of prevalence rate in Japan. Kasukawa R, Sharp G, eds. Mixed Connective Tissue Disease and Anti-nuclear Antibodies. Amsterdam: Excerpta Medica; 1987. 9.[↩][↩]
- Gunnarsson R, Molberg O, Gilboe IM, Gran JT; PAHNOR1 Study Group. The prevalence and incidence of mixed connective tissue disease: a national multicentre survey of Norwegian patients. Ann Rheum Dis. 2011 Jun;70(6):1047-51. doi: 10.1136/ard.2010.143792[↩][↩]
- Mixed Connective-Tissue Disease (MCTD). https://emedicine.medscape.com/article/335815-overview#a6[↩]
- Ferucci ED, Johnston JM, Gordon C, Helmick CG, Lim SS. Prevalence of Mixed Connective Tissue Disease in a Population-Based Registry of American Indian/Alaska Native People in 2007. Arthritis Care Res (Hoboken). 2017 Aug;69(8):1271-1275. doi: 10.1002/acr.23135[↩]
- Chauhan K, Surmachevska N, Hanna A. Relapsing Polychondritis. [Updated 2023 Jul 4]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK436007[↩][↩]
- Liu Y, Li X, Cheng L, Zhan H, Huang Y, Li H, Li Y. Progress and challenges in the use of blood biomarkers in relapsing polychondritis. Clin Exp Immunol. 2023 Jun 5;212(3):199-211. doi: 10.1093/cei/uxad014[↩]
- Sharma A, Gnanapandithan K, Sharma K, Sharma S. Relapsing polychondritis: a review. Clin Rheumatol. 2013 Nov;32(11):1575-83. doi: 10.1007/s10067-013-2328-x[↩]
- Butterton JR, Collier DS, Romero JM, Zembowicz A. Case records of the Massachusetts General Hospital. Case 14-2007. A 59-year-old man with fever and pain and swelling of both eyes and the right ear. N Engl J Med. 2007 May 10;356(19):1980-8. doi: 10.1056/NEJMcpc079009[↩]
- Borgia F, Giuffrida R, Guarneri F, Cannavò SP. Relapsing Polychondritis: An Updated Review. Biomedicines. 2018 Aug 2;6(3):84. doi: 10.3390/biomedicines6030084[↩]
- Masuda N, Nishikawa R, Ueda T, Ogata N. Severe panuveitis with relapsing polychondritis. Am J Ophthalmol Case Rep. 2018 Apr 20;11:3-5. doi: 10.1016/j.ajoc.2018.04.024[↩]
- Zheutlin A, Schiopu E. Relapsing Polychondritis following Treatment with Secukinumab for Ankylosing Spondylitis: Case Report and Review of the Literature. Case Rep Rheumatol. 2018 Jul 2;2018:6760806. doi: 10.1155/2018/6760806[↩]
- Neycheva S, Kamburova A. IL-17 Inhibitor Secukinumab Achieves Remission in Relapsing Polychondritis: A Case Report. Am J Case Rep. 2025 Mar 20;26:e946916. doi: 10.12659/AJCR.946916[↩]
- Winter G, Löffelmann T, Chaya S, Kaiser H, Prenzler NK, Warnecke A, Wetzke M, Derlin T, Renz D, Stueber T, Länger F, Schütz K, Schwerk N. Relapsing Polychondritis with Tracheobronchial Involvement: A Detailed Description of Two Pediatric Cases and Review of the Literature. Klin Padiatr. 2024 Feb;236(2):97-105. doi: 10.1055/a-2230-1521[↩]
- Arnaud L, Mathian A, Haroche J, et al. Pathogenesis of relapsing polychondritis: A 2013 update. Autoimmun Rev. 2014;13:90–95. doi: 10.1016/j.autrev.2013.07.005[↩]
- Trentham DE, Le CH. Relapsing polychondritis. Ann Intern Med. 1998;129:114–22. doi: 10.7326/0003-4819-129-2-199807150-00011[↩]
- D’Cruz DP, Ferrada MA. Relapsing Polychondritis and Large-vessel Vasculitis. J Rheumatol. 2020 Dec 1;47(12):1732-1733. doi: 10.3899/jrheum.200083[↩]
- Ostrowski RA, Takagishi T, Robinson J. Rheumatoid arthritis, spondyloarthropathies, and relapsing polychondritis. Handb Clin Neurol. 2014;119:449-61. doi: 10.1016/B978-0-7020-4086-3.00029-1[↩]
- Zeuner M, Straub RH, Rauh G, Albert ED, Schölmerich J, Lang B. Relapsing polychondritis: clinical and immunogenetic analysis of 62 patients. J Rheumatol. 1997 Jan;24(1):96-101.[↩]
- Dion J, Costedoat-Chalumeau N, Sène D, Cohen-Bittan J, Leroux G, Dion C, Francès C, Piette JC. Relapsing Polychondritis Can Be Characterized by Three Different Clinical Phenotypes: Analysis of a Recent Series of 142 Patients. Arthritis Rheumatol. 2016 Dec;68(12):2992-3001. doi: 10.1002/art.39790[↩]
- Horváth A, Páll N, Molnár K, et al. A nationwide study of the epidemiology of relapsing polychondritis. Clin Epidemiol. 2016;8:211–230. doi: 10.2147/CLEP.S91439[↩]
- Kent PD, Michet CJ, Luthra HS. Relapsing polychondritis. Curr Opin Rheumatol. 2004;16:56–61. doi: 10.1097/00002281-200401000-00011[↩]
- Jaksch-Wartenhorst R. Polychondropathia. Wien. Arch. Inn. Med. 1923;6:93–100.[↩]
- Pearson C.M., Kline H.M., Newcomer V.D. Relapsing polychondritis. N. Engl. J. Med. 1960 Jul 14;263:51-8. doi: 10.1056/NEJM196007142630201[↩][↩]
- Relapsing Polychondritis. https://emedicine.medscape.com/article/331475-overview#a7[↩]
- Relapsing Polychondritis. https://rarediseases.org/rare-diseases/relapsing-polychondritis/[↩][↩]
- Relapsing polychondritis in the Department of Defense population and review of the literature. Mathew SD, Battafarano DF, Morris MJ. Semin Arthritis Rheum. 2012 Aug; 42(1):70-83.[↩]
- Mertz, P., Costedoat-Chalumeau, N., Ferrada, M.A. et al. Relapsing polychondritis: clinical updates and new differential diagnoses. Nat Rev Rheumatol 20, 347–360 (2024). https://doi.org/10.1038/s41584-024-01113-9[↩]
- Relapsing polychondritis. Kent PD, Michet CJ Jr, Luthra HS. Curr Opin Rheumatol. 2004 Jan; 16(1):56-61.[↩]
- Incidence and mortality of relapsing polychondritis in the UK: a population-based cohort study. Hazra N, Dregan A, Charlton J, Gulliford MC, D’Cruz DP. Rheumatology (Oxford). 2015 Dec; 54(12):2181-7.[↩]
- Lekpa FK, Chevalier X. Refractory relapsing polychondritis: Challenges and solutions. Open Access Rheumatol. 2018;10:1–11. doi: 10.2147/OARRR.S142892[↩]
- Leung KK, Edani S. Red-eared zebra diagnosis: Case of relapsing polychondritis. Can Fam Physician. 2018 May;64(5):363-367. https://pmc.ncbi.nlm.nih.gov/articles/PMC5951651[↩]
- Kothari T, Valsamakis T, Sridhar A V et al. Case of paediatric relapsing polychondritis with severe airway involvement: the challenges of long-term airway and respiratory management. BMJ Case Rep. 2021;14:e239774. doi: 10.1136/bcr-239774[↩]
- Meshkov AD, Novikov PI, Zhilyaev EV, Ilevsky IDJ, Moiseev SV. Tofacitinib in steroid-dependent relapsing polychondritis. Ann. Rheum. Dis. 2018 May 03[↩]
- Moulis G, Pugnet G, Costedoat-Chalumeau N, Mathian A, Leroux G, Boutémy J, Espitia O, Bouillet L, Berthier S, Gaultier JB, Jeandel PY, Konaté A, Mékinian A, Solau-Gervais E, Terrier B, Wendling D, Andry F, Garnier C, Cathébras P, Arnaud L, Palmaro A, Cacoub P, Amoura Z, Piette JC, Arlet P, Lapeyre-Mestre M, Sailler L. Efficacy and safety of biologics in relapsing polychondritis: a French national multicentre study. Ann. Rheum. Dis. 2018 Aug;77(8):1172-1178[↩]
- Relapsing Polychondritis. N Engl J Med 2018; 378:1715 DOI: 10.1056/NEJMicm1713302 https://www.nejm.org/doi/full/10.1056/NEJMicm1713302[↩]
- Scleritis. https://eyerounds.org/atlas/pages/Scleritis13.htm#gsc.tab=0[↩]
- Carsons SE, Blum MA. Sjogren Syndrome. [Updated 2025 Jul 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK431049[↩][↩][↩][↩]
- Price EJ, Benjamin S, Bombardieri M, et al. British Society for Rheumatology guideline on management of adult and juvenile onset Sjögren disease. Rheumatology (Oxford). 2025 Feb 1;64(2):409-439. doi: 10.1093/rheumatology/keae152[↩]
- Ma D, Feng Y, Lin X. Immune and non-immune mediators in the fibrosis pathogenesis of salivary gland in Sjögren’s syndrome. Front Immunol. 2024 Oct 14;15:1421436. doi: 10.3389/fimmu.2024.1421436[↩]
- Meng Q, Ma J, Cui J, Gu Y, Shan Y. Subpopulation dynamics of T and B lymphocytes in Sjögren’s syndrome: implications for disease activity and treatment. Front Immunol. 2024 Sep 3;15:1468469. doi: 10.3389/fimmu.2024.1468469[↩]
- Yura Y, Hamada M. Outline of Salivary Gland Pathogenesis of Sjögren’s Syndrome and Current Therapeutic Approaches. Int J Mol Sci. 2023 Jul 6;24(13):11179. doi: 10.3390/ijms241311179[↩]
- Yayla ME, Aksoy R, Uslu E, Karaman Z, Yilmaz R, Ilbay A, Göveç Giynaş N, Gaydan A, Usta A, Mahmutoğlu Y, Ateş A, Turgay M. Are there differences in the clinical and laboratory features of patients with seronegative primary Sjögren’s syndrome? Turk J Med Sci. 2024 Oct 7;54(5):956-962. doi: 10.55730/1300-0144.5873[↩]
- Shiboski CH, Shiboski SC, Seror R, Criswell LA, Labetoulle M, Lietman TM, Rasmussen A, Scofield H, Vitali C, Bowman SJ, Mariette X; International Sjögren’s Syndrome Criteria Working Group. 2016 American College of Rheumatology/European League Against Rheumatism Classification Criteria for Primary Sjögren’s Syndrome: A Consensus and Data-Driven Methodology Involving Three International Patient Cohorts. Arthritis Rheumatol. 2017 Jan;69(1):35-45. doi: 10.1002/art.39859[↩]
- Baldini C, Ferro F, Bombardieri S. Classification criteria in Sjögren’s syndrome. Ann Transl Med. 2017 Aug;5(15):313. doi: 10.21037/atm.2017.05.07[↩][↩]
- Brito-Zerón P, Baldini C, Bootsma H, Bowman SJ, Jonsson R, Mariette X, Sivils K, Theander E, Tzioufas A, Ramos-Casals M. Sjögren syndrome. Nat Rev Dis Primers. 2016 Jul 7;2:16047. doi: 10.1038/nrdp.2016.47[↩]
- Brito-Zeron P, Theander E, Baldini C, Seror R, Retamozo S, et al. Early diagnosis of primary Sjogren’s syndrome: EULAR-SS task force clinical recommendations. Expert Review of Clinical Immunology. 2016;12:137–156. doi: 10.1586/1744666X.2016.1109449[↩]
- Mihai A, Caruntu C, Jurcut C, Blajut FC, Casian M, Opris-Belinski D, Ionescu R, Caruntu A. The Spectrum of Extraglandular Manifestations in Primary Sjögren’s Syndrome. J Pers Med. 2023 Jun 7;13(6):961. doi: 10.3390/jpm13060961[↩]
- Kiadaliri AA, Mohammad AJ, Englund M. Hospitalizations due to systemic connective tissue diseases: Secular trends and regional disparities in Sweden, 1998-2016. Int J Rheum Dis. 2018 Nov;21(11):1900-1906. doi: 10.1111/1756-185X.13341[↩]
- Alani H, Henty JR, Thompson NL, Jury E, Ciurtin C. Systematic review and meta-analysis of the epidemiology of polyautoimmunity in Sjögren’s syndrome (secondary Sjögren’s syndrome) focusing on autoimmune rheumatic diseases. Scand J Rheumatol. 2018 Mar;47(2):141-154. doi: 10.1080/03009742.2017.1324909[↩]
- Ramos-Casals M, Brito-Zerón P, Kostov B, Sisó-Almirall A, Bosch X, Buss D, et al. Google-driven search for big data in autoimmune geoepidemiology: analysis of 394,827 patients with systemic autoimmune diseases. Autoimmun Rev. (2015) 14:670–9. doi: 10.1016/j.autrev.2015.03.008[↩]
- Chivasso C., Sarrand J., Perret J., Delporte C., Soyfoo M.S. The Involvement of Innate and Adaptive Immunity in the Initiation and Perpetuation of Sjögren’s Syndrome. Int. J. Mol. Sci. 2021;22:658. doi: 10.3390/ijms22020658[↩]
- Baldini C, Fulvio G, La Rocca G, Ferro F. Update on the pathophysiology and treatment of primary Sjögren syndrome. Nat Rev Rheumatol. 2024 Aug;20(8):473-491. doi: 10.1038/s41584-024-01135-3[↩]
- Lessard CJ, Li H, Adrianto I, et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet. 2013 Nov;45(11):1284-92. doi: 10.1038/ng.2792[↩][↩]
- Baer AN, Akpek EK, Alevizos I; 18-21 April 2018, Washington, DC, USA. 14th International Symposium on Sjögren’s Syndrome. Clin Exp Rheumatol. 2018 May-Jun;36 Suppl 112(3):241-255. https://www.clinexprheumatol.org/abstract.asp?a=13333[↩]
- Kroese FGM, Haacke EA, Bombardieri M. The role of salivary gland histopathology in primary Sjögren’s syndrome: promises and pitfalls. Clin Exp Rheumatol. 2018 May-Jun;36 Suppl 112(3):222-233. https://www.clinexprheumatol.org/abstract.asp?a=13124[↩]
- Björk A, Mofors J, Wahren-Herlenius M. Environmental factors in the pathogenesis of primary Sjögren’s syndrome. J Intern Med. 2020 May;287(5):475-492. doi: 10.1111/joim.13032[↩]
- Bunya VY, Fernandez KB, Ying GS, Massaro-Giordano M, Macchi I, Sulewski ME, Hammersmith KM, Nagra PK, Rapuano CJ, Orlin SE. Survey of Ophthalmologists Regarding Practice Patterns for Dry Eye and Sjogren Syndrome. Eye Contact Lens. 2018 Nov;44 Suppl 2(Suppl 2):S196-S201. doi: 10.1097/ICL.0000000000000448[↩]
- Kittridge A, Routhouska SB, Korman NJ. Dermatologic Manifestations of Sjögren Syndrome. Journal of Cutaneous Medicine and Surgery. 2011;15(1):8-14. doi:10.2310/7750.2010.09033[↩]
- Sjogren Syndrome. https://emedicine.medscape.com/article/332125-overview[↩]
- Miklovic T, Sieg VC. Ehlers-Danlos Syndrome. [Updated 2023 May 29]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549814[↩]
- Malfait F, Francomano C, Byers P, Belmont J, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017 Mar;175(1):8-26. doi: 10.1002/ajmg.c.31552[↩][↩]
- EDS Types. https://www.ehlers-danlos.com/eds-types/#chart[↩]
- Dietz H. FBN1-Related Marfan Syndrome. 2001 Apr 18 [Updated 2022 Feb 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1335[↩][↩]
- Salik I, Rawla P. Marfan Syndrome. [Updated 2023 Jan 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537339[↩][↩][↩][↩]
- Marfan syndrome. https://medlineplus.gov/genetics/condition/marfan-syndrome[↩][↩][↩][↩][↩]
- Marfan Syndrome. https://rarediseases.org/rare-diseases/marfan-syndrome[↩]
- Marfan Syndrome. https://marfan.org/conditions/marfan-syndrome[↩]
- Milewicz DM, Braverman AC, De Backer J, Morris SA, Boileau C, Maumenee IH, Jondeau G, Evangelista A, Pyeritz RE. Marfan syndrome. Nat Rev Dis Primers. 2021 Sep 2;7(1):64. doi: 10.1038/s41572-021-00298-7. Erratum in: Nat Rev Dis Primers. 2022 Jan 17;8(1):3. doi: 10.1038/s41572-022-00338-w[↩]
- Muiño-Mosquera L, Cervi E, De Groote K, Dewals W, Fejzic Z, Kazamia K, Mathur S, Milleron O, Mir TS, Nielsen DG, Odermarsky M, Sabate-Rotes A, van der Hulst A, Valenzuela I, Jondeau G. Management of aortic disease in children with FBN1-related Marfan syndrome. Eur Heart J. 2024 Oct 14;45(39):4156-4169. doi: 10.1093/eurheartj/ehae526[↩]
- Edouard T, Picot MC, Bajanca F, Huguet H, Guitarte A, Langeois M, Chesneau B, Van Kien PK, Garrigue E, Dulac Y, Amedro P. Health-related quality of life in children and adolescents with Marfan syndrome or related disorders: a controlled cross-sectional study. Orphanet J Rare Dis. 2024 Apr 30;19(1):180. doi: 10.1186/s13023-024-03191-0[↩]
- Taniguchi Y, Takeda N, Inuzuka R, Matsubayashi Y, Kato S, Doi T, Yagi H, Yamauchi H, Ando M, Oshima Y, Tanaka S. Impact of pathogenic FBN1 variant types on the development of severe scoliosis in patients with Marfan syndrome. J Med Genet. 2023 Jan;60(1):74-80. doi: 10.1136/jmedgenet-2021-108186[↩]
- Laganà G, Venza N, Malara A, Liguori C, Cozza P, Pisano C. Obstructive Sleep Apnea, Palatal Morphology, and Aortic Dilatation in Marfan Syndrome Growing Subjects: A Retrospective Study. Int J Environ Res Public Health. 2021 Mar 16;18(6):3045. doi: 10.3390/ijerph18063045[↩]
- Trawicka A, Lewandowska-Walter A, Majkowicz M, Sabiniewicz R, Woźniak-Mielczarek L. Health-Related Quality of Life of Patients with Marfan Syndrome-Polish Study. Int J Environ Res Public Health. 2022 Jun 2;19(11):6827. doi: 10.3390/ijerph19116827[↩]
- Pollock L, Ridout A, Teh J, Nnadi C, Stavroulias D, Pitcher A, Blair E, Wordsworth P, Vincent TL. The Musculoskeletal Manifestations of Marfan Syndrome: Diagnosis, Impact, and Management. Curr Rheumatol Rep. 2021 Nov 26;23(11):81. doi: 10.1007/s11926-021-01045-3[↩]
- Du Q, Zhang D, Zhuang Y, Xia Q, Wen T, Jia H. The Molecular Genetics of Marfan Syndrome. Int J Med Sci. 2021 May 27;18(13):2752-2766. doi: 10.7150/ijms.60685[↩]
- Baban A, Parlapiano G, Cicenia M, Armando M, Franceschini A, Pacifico C, Panfili A, Zinzanella G, Romanzo A, Fusco A, Caiazza M, Perri G, Galletti L, Digilio MC, Buonuomo PS, Bartuli A, Novelli A, Raponi M, Limongelli G. Unique Features of Cardiovascular Involvement and Progression in Children with Marfan Syndrome Justify Dedicated Multidisciplinary Care. J Cardiovasc Dev Dis. 2024 Apr 3;11(4):114. doi: 10.3390/jcdd11040114[↩]
- Zeigler SM, Sloan B, Jones JA. Pathophysiology and Pathogenesis of Marfan Syndrome. Adv Exp Med Biol. 2021;1348:185-206. doi: 10.1007/978-3-030-80614-9_8[↩]
- Andersen NH, Hauge EM, Baad-Hansen T, Groth KA, Berglund A, Gravholt CH, Stochholm K. Musculoskeletal diseases in Marfan syndrome: a nationwide registry study. Orphanet J Rare Dis. 2022 Mar 5;17(1):118. doi: 10.1186/s13023-022-02272-2[↩]
- Zodanu GKE, Hwang JH, Mehta Z, Sisniega C, Barsegian A, Kang X, Biniwale R, Si MS, Satou GM, Halnon N, Ucla Congenital Heart Defect BioCore Faculty, Grody WW, Van Arsdell GS, Nelson SF, Touma M. High-Throughput Genomics Identify Novel FBN1/2 Variants in Severe Neonatal Marfan Syndrome and Congenital Heart Defects. Int J Mol Sci. 2024 May 17;25(10):5469. doi: 10.3390/ijms25105469[↩]
- von Kodolitsch Y, Robinson PN. Marfan syndrome: an update of genetics, medical and surgical management. Heart. 2007 Jun;93(6):755-60. doi: 10.1136/hrt.2006.098798[↩]
- Isselbacher EM, Preventza O, Hamilton Black J, Augoustides JG, Beck AW, Bolen MA et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022;146:e334–482. 10.1161/CIR.0000000000001106[↩]
- Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwöger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ; ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014 Nov 1;35(41):2873-926. doi: 10.1093/eurheartj/ehu281. Epub 2014 Aug 29. Erratum in: Eur Heart J. 2015 Nov 1;36(41):2779. doi: 10.1093/eurheartj/ehv178[↩]
- Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005 Nov 15;139C(1):4-9. doi: 10.1002/ajmg.c.30068[↩]
- Hilhorst-Hofstee Y, Rijlaarsdam ME, Scholte AJ, Swart-van den Berg M, Versteegh MI, van der Schoot-van Velzen I, Schäbitz HJ, Bijlsma EK, Baars MJ, Kerstjens-Frederikse WS, Giltay JC, Hamel BC, Breuning MH, Pals G. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat. 2010 Dec;31(12):E1915-27. doi: 10.1002/humu.21372[↩]
- Sakai LY, Keene DR, Glanville RW, Bächinger HP. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J Biol Chem. 1991 Aug 5;266(22):14763-70. https://www.jbc.org/article/S0021-9258(18)98752-1/pdf[↩]
- Dietz HC, Ramirez F, Sakai LY. Marfan’s syndrome and other microfibrillar diseases. Adv Hum Genet. 1994;22:153-86. doi: 10.1007/978-1-4757-9062-7_4[↩]
- FBN1 gene. https://medlineplus.gov/genetics/gene/fbn1[↩]
- Reinhardt DP, Keene DR, Corson GM, Pöschl E, Bächinger HP, Gambee JE, Sakai LY. Fibrillin-1: organization in microfibrils and structural properties. J Mol Biol. 1996 Apr 26;258(1):104-16. doi: 10.1006/jmbi.1996.0237[↩]
- Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995 May;129(4):1165-76. https://pmc.ncbi.nlm.nih.gov/articles/instance/2120487/pdf/jc12941165.pdf[↩]
- Ammash NM, Sundt TM, Connolly HM. Marfan syndrome-diagnosis and management. Curr Probl Cardiol. 2008 Jan;33(1):7-39. doi: 10.1016/j.cpcardiol.2007.10.001[↩]
- LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011 Feb;8(2):103-13. doi: 10.1038/nrcardio.2010.187[↩]
- Loeys BL, Dietz HC. Loeys-Dietz Syndrome. 2008 Feb 28 [Updated 2024 Sep 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1133[↩][↩][↩][↩][↩][↩]
- About Loeys-Dietz Syndrome. https://www.loeysdietz.org/en/medical-information[↩][↩][↩]
- Loeys-Dietz Syndrome. https://rarediseases.org/rare-diseases/loeys-dietz-syndrome[↩][↩][↩]
- Ziegler A, Duclaux-Loras R, Revenu C, et al. Bi-allelic variants in IPO8 cause a connective tissue disorder associated with cardiovascular defects, skeletal abnormalities, and immune dysregulation. Am J Hum Genet. 2021 Jun 3;108(6):1126-1137. doi: 10.1016/j.ajhg.2021.04.020[↩]
- Loeys BL, Dietz HC. Loeys-Dietz Syndrome. 2008 Feb 28 [Updated 2018 Mar 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1133[↩][↩]
- Lynch CP, Patel M, Seeley AH, Seeley MA. Orthopaedic Management of Loeys-Dietz Syndrome: A Systematic Review. J Am Acad Orthop Surg Glob Res Rev. 2021 Nov 15;5(11):e21.00087. doi: 10.5435/JAAOSGlobal-D-21-00087[↩]
- Mortier G. Stickler Syndrome. 2000 Jun 9 [Updated 2023 Sep 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1302[↩][↩]
- Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. 2000 Jun 9 [Updated 2017 Mar 16]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1302/[↩][↩]
- Acke FRE, De Leenheer EMR. Hearing Loss in Stickler Syndrome: An Update. Genes (Basel). 2022 Sep 1;13(9):1571. doi: 10.3390/genes13091571[↩]
- Fernández-Pérez JJ, Mascaraque-Ruiz P, Martín Gómez C, Martínez-Caballero I, Otón T, Carmona L, Lara SL. Quality of Life in Children and Adolescents with Stickler Syndrome in Spain. Children (Basel). 2022 Aug 19;9(8):1255. doi: 10.3390/children9081255[↩]
- Stickler G.B., Hughes W., Houchin P. Clinical Features of Hereditary Progressive Arthro-Ophthalmopathy (Stickler Syndrome): A Survey. Genet. Med. 2001;3:192–196. doi: 10.1097/00125817-200105000-00008[↩]
- Stickler G., Belau P., Farrell F., Jones J., Pugh D., Steinberg A., Ward L. Hereditary Progessive Arthro-Ophthalmopathy. Mayo Clin. Proc. 1965;13:433–455. doi: 10.1017/s1120962300015900[↩][↩]
- Rose P.S., Levy H.P., Liberfarb R.M., Davis J., Szymko-Bennett Y., Rubin B.I., Tsilou E., Griffith A.J., Francomano C.A. Stickler Syndrome: Clinical Characteristics and Diagnostic Criteria. Am. J. Med. Genet. Part A. 2005;138:199–207. doi: 10.1002/ajmg.a.30955[↩][↩]
- Webb A.C., Markus A.F. The Diagnosis and Consequences of Stickler Syndrome. Br. J. Oral Maxillofac. Surg. 2002;40:49–51. doi: 10.1054/bjom.2001.0747[↩][↩]
- Alexander P, Fincham GS, Brown S, Collins D, McNinch AM, Poulson AV, Richards A, Martin H, Wareham N, Snead MP. Cambridge Prophylactic Protocol, Retinal Detachment, and Stickler Syndrome. N Engl J Med. 2023 Apr 6;388(14):1337-1339. doi: 10.1056/NEJMc2211320[↩]
- Karempelis P, Hagen M, Morrell N, Roby BB. Associated syndromes in patients with Pierre Robin Sequence. Int J Pediatr Otorhinolaryngol. 2020 Apr;131:109842. doi: 10.1016/j.ijporl.2019.109842[↩]
- Davies A, Davies A, Wren Y, Deacon S, Cobb A, McLean N, David D, Chummun S. Syndromes associated with Robin sequence: a national prospective cohort study. Arch Dis Child. 2023 Jan;108(1):42-46. doi: 10.1136/archdischild-2022-324722[↩]
- Snead M, Martin H, Bale P, Shenker N, Baguley D, Alexander P, McNinch A, Poulson A. Therapeutic and diagnostic advances in Stickler syndrome. Ther Adv Rare Dis. 2020 Dec 9;1:2633004020978661. doi: 10.1177/2633004020978661[↩][↩][↩]
- Hoornaert KP, Vereecke I, Dewinter C, et al. Stickler syndrome caused by COL2A1 mutations: genotype-phenotype correlation in a series of 100 patients. Eur J Hum Genet. 2010 Aug;18(8):872-80. doi: 10.1038/ejhg.2010.23. Erratum in: Eur J Hum Genet. 2010 Aug;18(8):881.[↩][↩]
- Boothe M, Morris R, Robin N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. J Pers Med. 2020 Aug 27;10(3):105. doi: 10.3390/jpm10030105[↩]
- Wang DD, Gao FJ, Hu FY, Zhang SH, Xu P, Wu JH. Mutation Spectrum of Stickler Syndrome Type I and Genotype-phenotype Analysis in East Asian Population: a systematic review. BMC Med Genet. 2020 Feb 10;21(1):27. doi: 10.1186/s12881-020-0963-z[↩][↩]
- Snead MP, Yates JR. Clinical and Molecular genetics of Stickler syndrome. J Med Genet. 1999 May;36(5):353-9. https://pmc.ncbi.nlm.nih.gov/articles/instance/1734362/pdf/v036p00353.pdf[↩]
- Richards A.J., Baguley D.M., Yates J.R.W., Lane C., Nicol M., Harper P.S., Scott J.D., Snead M.P. Variation in the Vitreous Phenotype of Stickler Syndrome Can Be Caused by Different Amino Acid Substitutions in the X Position of the Type II Collagen Gly-X-Y Triple Helix. Am. J. Hum. Genet. 2000;67:1083–1094. doi: 10.1016/S0002-9297(07)62938-3[↩][↩][↩]
- Acke F.R., Swinnen F.K., Malfait F., Dhooge I.J., De Leenheer E.M.R. Auditory Phenotype in Stickler Syndrome: Results of Audiometric Analysis in 20 Patients. Eur. Arch. Otorhinolaryngol. 2016;273:3025–3034. doi: 10.1007/s00405-016-3896-6[↩][↩][↩]
- Snead M.P., McNinch A.M., Poulson A.V., Bearcroft P., Silverman B., Gomersall P., Parfect V., Richards A.J. Stickler Syndrome, Ocular-Only Variants and a Key Diagnostic Role for the Ophthalmologist. Eye. 2011;25:1389. doi: 10.1038/eye.2011.201[↩][↩][↩]
- Stickler Syndrome. https://rarediseases.org/rare-diseases/stickler-syndrome[↩][↩]
- Printzlau A, Andersen M. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study. Cleft Palate Craniofac J. 2004;41:47–52. https://www.ncbi.nlm.nih.gov/pubmed/14697070[↩]
- Richards AJ, McNinch A, Martin H, Oakhill K, Rai H, Waller S, Treacy B, Whittaker J, Meredith S, Poulson A, Snead MP. Stickler syndrome and the vitreous phenotype: mutations in COL2A1 and COL11A1. Hum Mutat. 2010 Jun;31(6):E1461-71. doi: 10.1002/humu.21257[↩]
- What Is a Soft Tissue Sarcoma? https://www.cancer.org/cancer/types/soft-tissue-sarcoma/about/soft-tissue-sarcoma.html[↩][↩]
- The Voice of 12,000 Patients. https://www.eurordis.org/wp-content/uploads/2009/12/EURORDISCARE_FULLBOOKr.pdf[↩]
- Clark S. Help me trust you after my misdiagnosis. BMJ. 2021 May 26;373:n1175. doi: 10.1136/bmj.n1175[↩]
- Halverson CME, Clayton EW, Garcia Sierra A, Francomano C. Patients with Ehlers-Danlos syndrome on the diagnostic odyssey: Rethinking complexity and difficulty as a hero’s journey. Am J Med Genet C Semin Med Genet. 2021 Dec;187(4):416-424. doi: 10.1002/ajmg.c.31935[↩]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Care Services; Committee on Selected Heritable Disorders of Connective Tissue and Disability; Wedge RA, Cartaxo T, Spicer CM, et al., editors. Selected Heritable Disorders of Connective Tissue and Disability. Washington (DC): National Academies Press (US); 2022 Jul 8. 2, Overview of Hereditary Disorders of Connective Tissue. Available from: https://www.ncbi.nlm.nih.gov/books/NBK584965[↩]
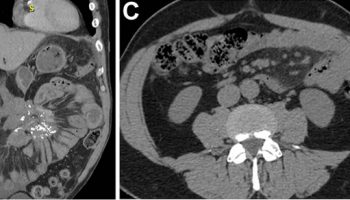
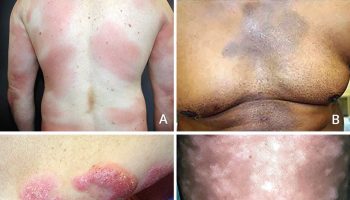
{kind=link}