Contents
CPEO disease
CPEO is an acronym for chronic progressive external ophthalmoplegia and CPEO is also called progressive external ophthalmoplegia (PEO), is a inherited mitochondrial disorder or POLG-related disorder that is characterized by weakness or paralysis of the eye muscles that control eye movement (extraocular muscles) and eyelids, leading to drooping eyelids (ptosis) and difficulty moving the eyes (ophthalmoplegia) 1, 2, 3, 4, 5, 6, 7, 8, 9, 10), 11, 12, 13, 14, 13, 15, 16. As the name suggests, CPEO (chronic progressive external ophthalmoplegia) typically progresses slowly over time (chronic), with symptoms worsening gradually. CPEO (chronic progressive external ophthalmoplegia) typically appears in adults between ages 18 and 40 and slowly worsens over time. The first sign of CPEO (chronic progressive external ophthalmoplegia) is typically drooping eyelids (ptosis), which can affect one or both eyelids. As drooping eyelid (ptosis) worsens, affected individuals may use the forehead muscles to try to lift the eyelids, or they may lift up their chin in order to see. However, there are cases, although rare, that can present without drooping eyelid (ptosis) in the adult life making CPEO diagnosis challenging 17. Another characteristic feature of CPEO (chronic progressive external ophthalmoplegia) is weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Affected individuals have to turn their head to see in different directions, especially as the ophthalmoplegia worsens. People with CPEO (chronic progressive external ophthalmoplegia) may also have general weakness of the muscles used for movement (myopathy), particularly those in the neck, arms, or legs. The weakness may be especially noticeable during exercise (exercise intolerance). Muscle weakness may also cause difficulty swallowing (dysphagia).
Although muscle weakness is the primary symptom of CPEO (chronic progressive external ophthalmoplegia), this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as chronic progressive external ophthalmoplegia plus (CPEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
Drooping eyelid (ptosis) and weakness or paralysis of the muscles that move the eye (ophthalmoplegia) are nonspecific signs and symptoms and can be mimicked by other nerve damage (neuropathic), neuromuscular junction, and musclular (myogenic) causes. CPEO (chronic progressive external ophthalmoplegia) can sometimes difficult to diagnose however, especially when the patient eye condition is asymmetric (affecting one eye only), lacks other other signs and symptoms, or has no family history of CPEO 10). Therefore, it is crucial for your eye doctor to have CPEO (chronic progressive external ophthalmoplegia) in his/her list of possible diagnoses to avoid unnecessary tests or delays in diagnosis 18.
The worldwide prevalence of CPEO is unknown; however, the incidence of CPEO is 1 per 100,000 to 2 per 100,000 3. In the United Kingdom, the estimated prevalence of CPEO recorded was 1 in 30,000 19.
CPEO can result from mutations in one of several different genes. In some CPEO cases, mutations occur in the POLG, TWNK, RRM2B, and SLC25A4 genes 20, 11. These genes (i.e., POLG, TWNK, RRM2B, and SLC25A4) are critical for the production and maintenance of mitochondrial DNA (mtDNA). Less commonly, mutations that change single nucleotides in genes found in mitochondrial DNA (mtDNA), such as the MT-TL1 gene, cause chronic progressive external ophthalmoplegia (CPEO). These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with CPEO (chronic progressive external ophthalmoplegia) are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
There are at least 67 POLG gene mutations that cause chronic progressive external ophthalmoplegia (CPEO). Gene that has been associated with CPEO include 21, 22:
- POLG
- POLG2
- TK2
- OPA1
- RRM2B
- TWNK
- SLC25A
- RRM1
- TOP3A
- C1QBP
- DNA2
- C10ORF2
- DGUOK
- MPV17
- MGME1
- SPG7
- AFG3L2
- RNASEH1
- GMPR
Most POLG gene mutations change single amino acids in the alpha subunit of polymerase gamma (pol γ), which decreases the efficiency of mitochondrial DNA (mtDNA) replication. As in another POLG-related disorder, the most common POLG gene mutation in CPEO (chronic progressive external ophthalmoplegia) is Ala467Thr (amino acid alanine is replaced with the amino acid threonine at position 467). Ala467Thr mutation blocks the ability of the alpha subunit to attach to the beta subunits and reduces polymerase gamma (pol γ) ability to synthesize DNA. Although the mechanism is unknown, up to 60% cases of mitochondrial CPEO are due to mitochondrial DNA (mtDNA) deletions or have fewer copies of mtDNA (mtDNA depletion) 11. This abnormality (mtDNA depletion) is seen only in the tissues affected by the disease. MtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy. Researchers have not determined how deletions of mtDNA lead to the specific signs and symptoms of chronic progressive external ophthalmoplegia (CPEO), although the features of the condition are probably related to impaired oxidative phosphorylation 23. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
Enzymes encoded by mitochondrial DNA (mtDNA) play a key role in oxidative phosphorylation necessary to meet the metabolic demands of active muscle 24. To maintain a high fatigue resistance, eye muscles that control eye movement (extraocular muscles) have adapted to have both a higher mitochondrial content and higher metabolic demand compared to skeletal muscles (the most common type of muscle in your body which is connected to your bones to form part of the skeletal system which moves your limbs and other parts of your body). These properties are hypothesized to be one of the reasons why eye muscles that control eye movement (extraocular muscles) are especially vulnerable to the oxidative phosphorylation dysfunction that occurs in CPEO (chronic progressive external ophthalmoplegia) 24.
Because CPEO (chronic progressive external ophthalmoplegia) is a inherited genetic disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
There is no cure for CPEO (chronic progressive external ophthalmoplegia), but treatment focuses on managing symptoms and improving quality of life. Chronic progressive external ophthalmoplegia (CPEO) treatment may include physical therapy, medications to address specific symptoms like ptosis, and support for other affected organ systems.
Figure 1. CPEO (Chronic Progressive External Ophthalmoplegia)
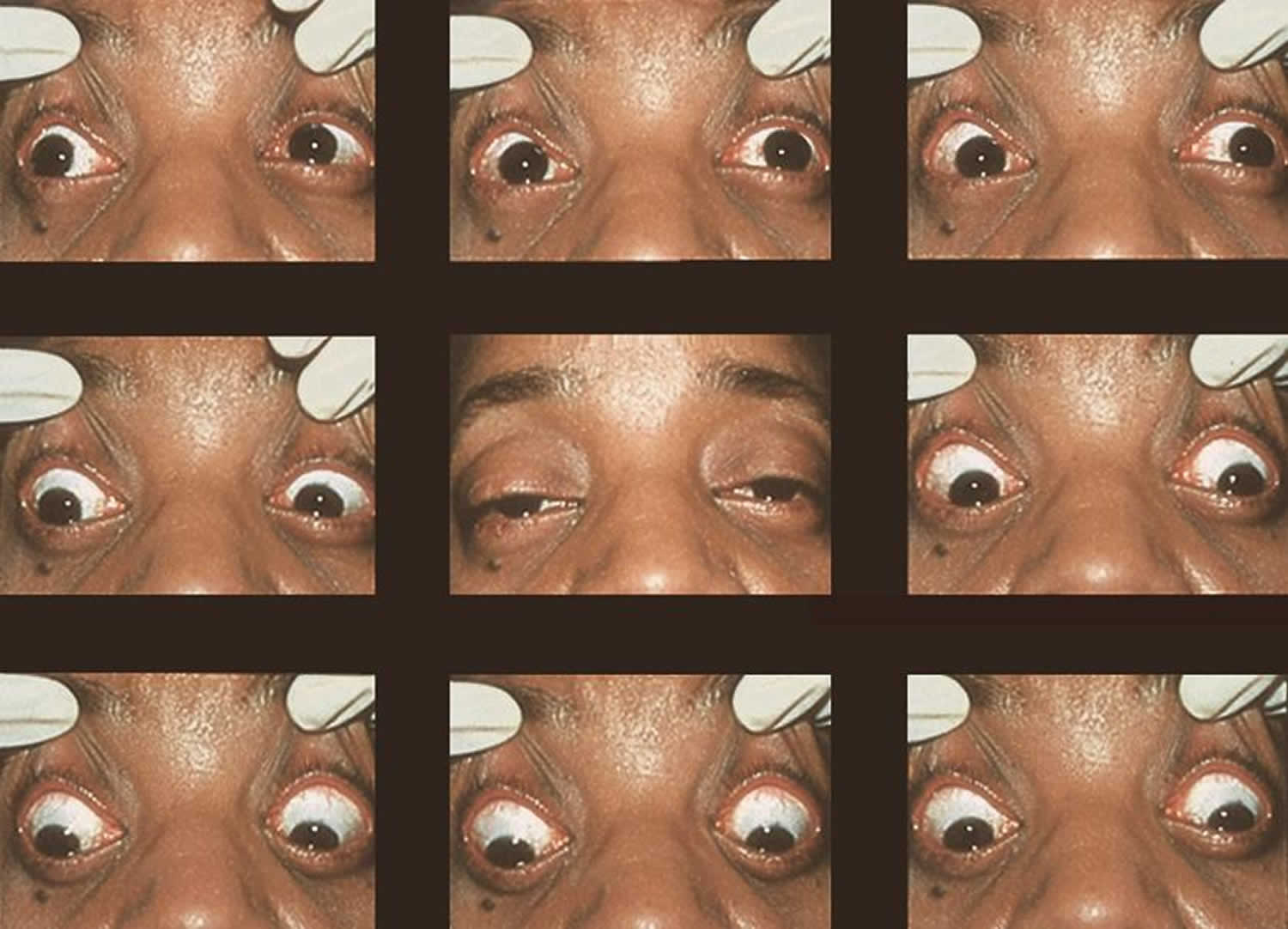
Footnotes: In its isolated form, CPEO is typically a sporadic disorder characterized by progressive bilateral progressive drooping eyelid (ptosis) and weakness or paralysis of the muscles that move the eye (ophthalmoplegia). A 42-year-old with a 2-year history of progressive drooping eyelid (ptosis) and weakness or paralysis of the muscles that move the eye (ophthalmoplegia). Biopsy of the deltoid muscle showed ragged red fibers consistent with CPEO (chronic progressive external ophthalmoplegia).
[Source 10) ]Figure 2. CPEO Plus (CPEO+)
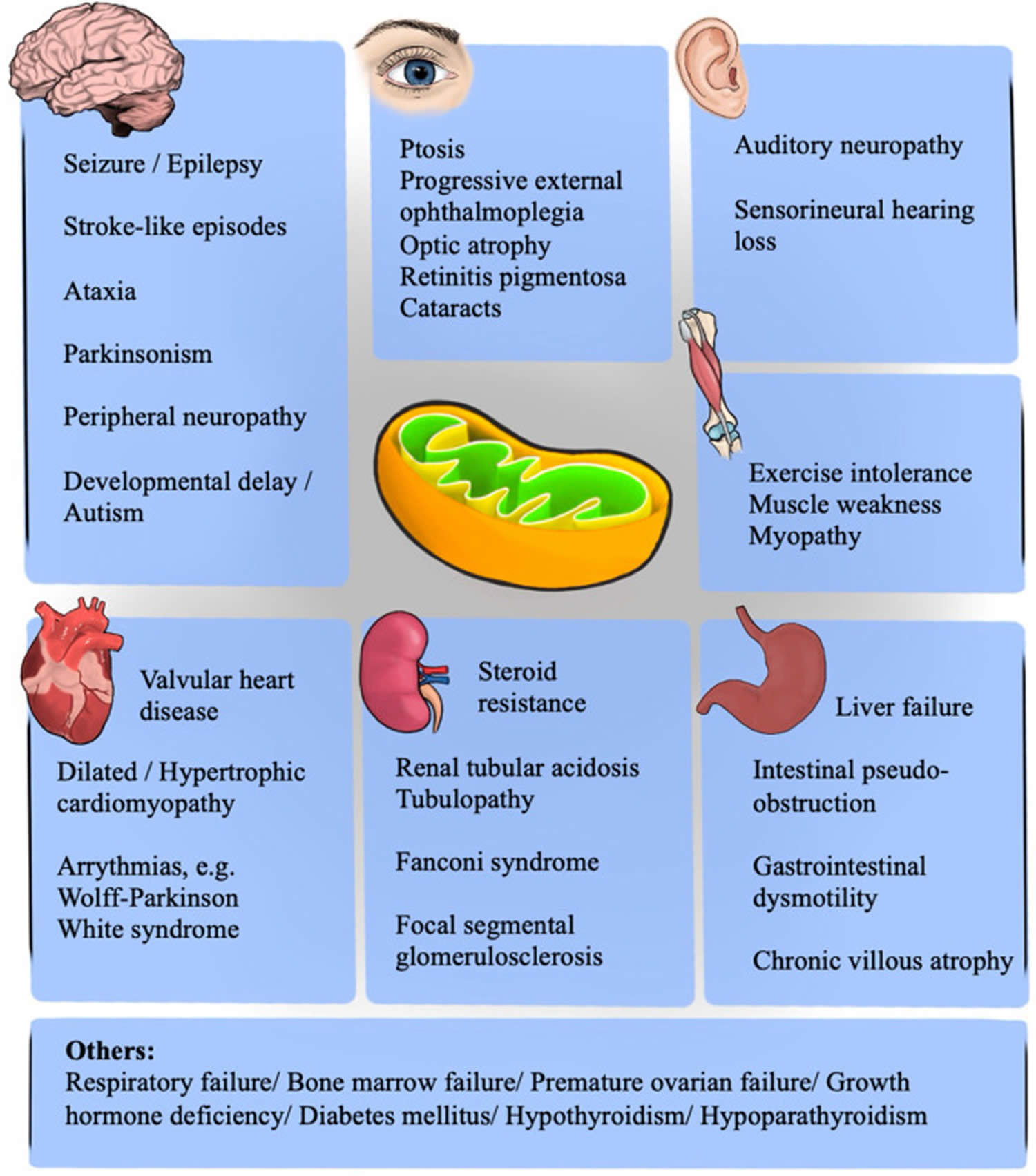
What is mitochondria?
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called adenosine triphosphate (ATP) that your cells can use 25. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP (adenosine triphosphate). Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus.
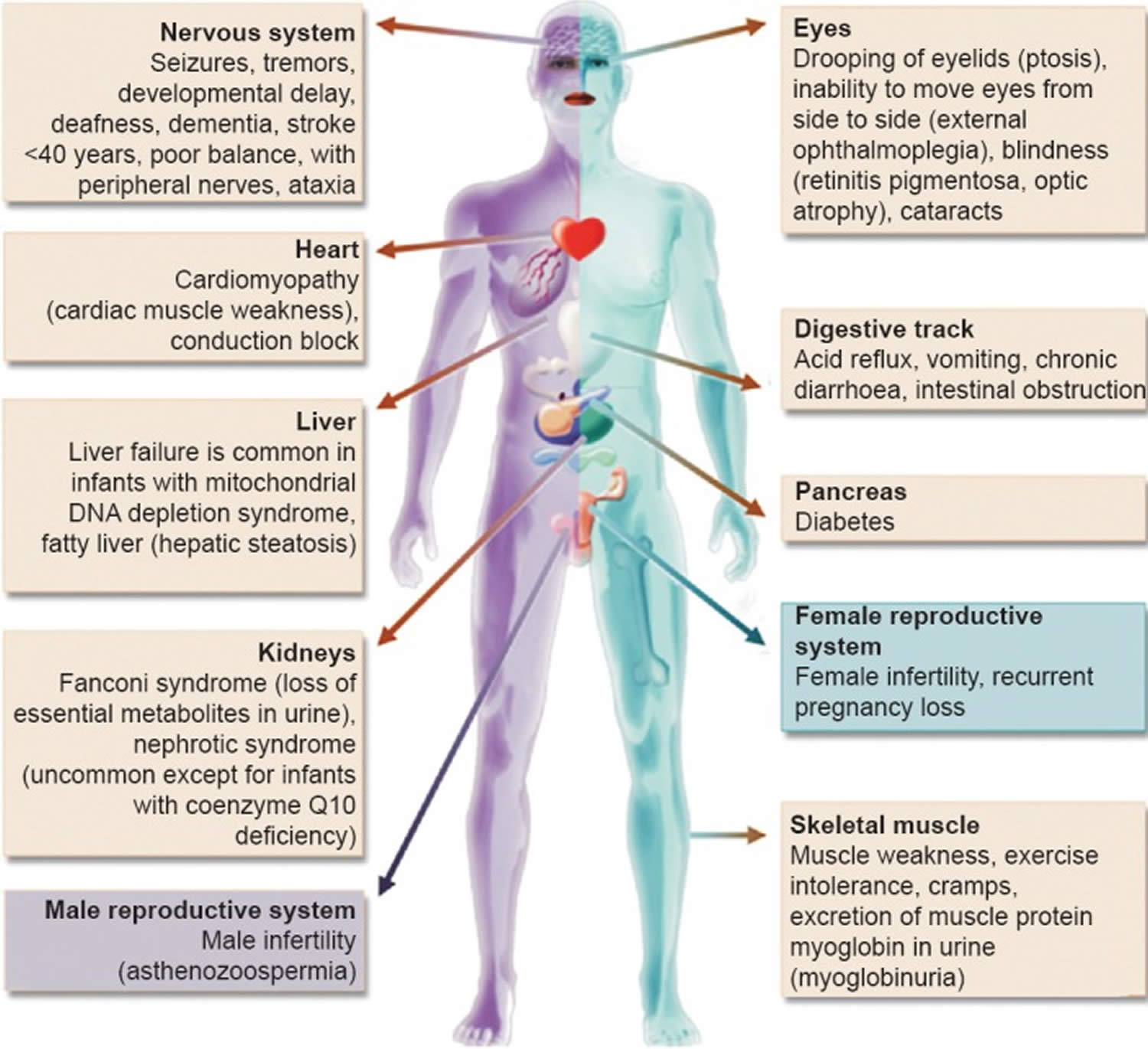
The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. The parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 26. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 27. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 3. Mitochondria cell
What is mitochondrial disease?
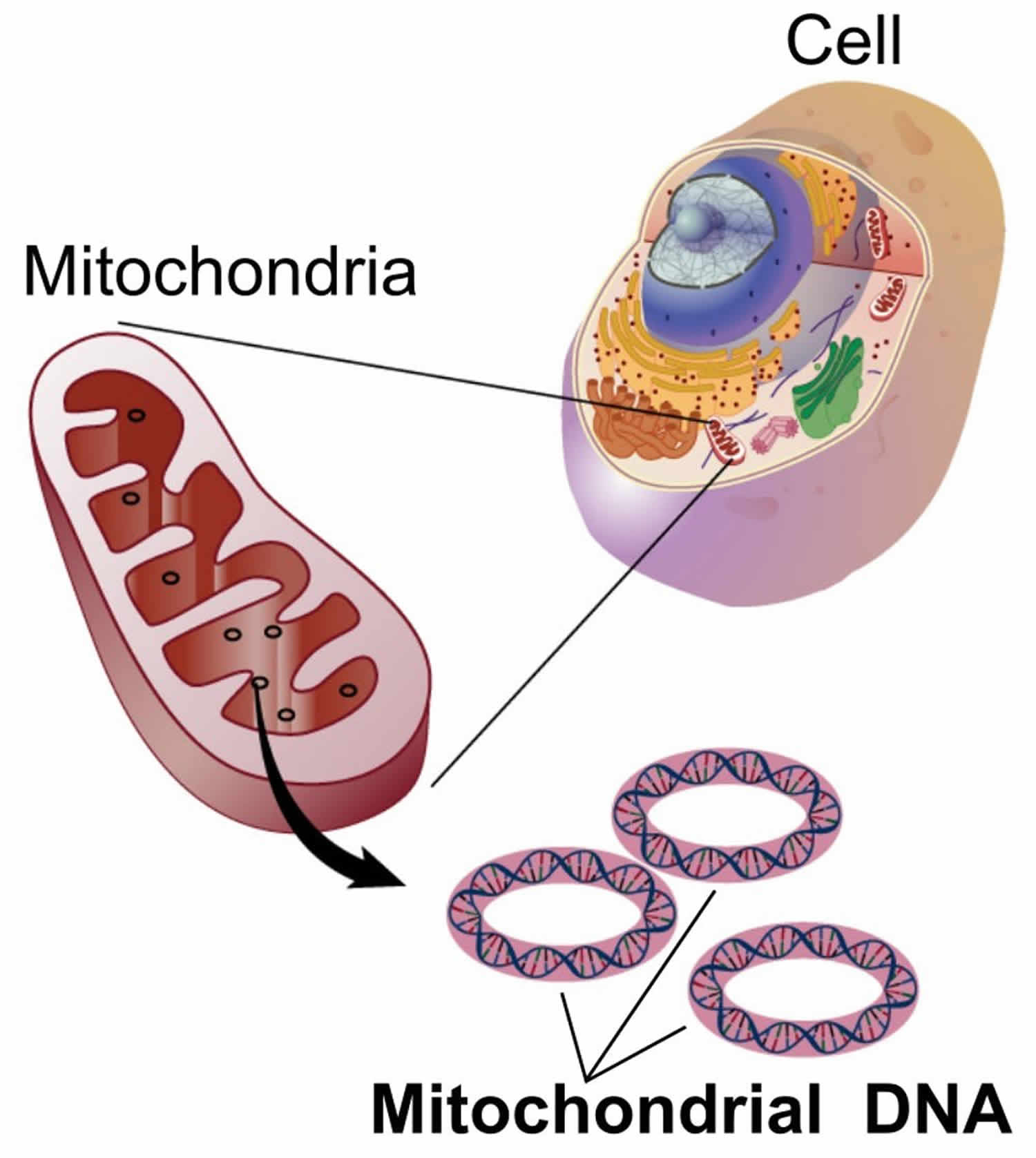
Mitochondrial diseases can affect almost any part of your body, including the cells of your:
- Brain.
- Nerves.
- Muscles.
- Kidneys.
- Heart.
- Liver.
- Eyes.
- Ears.
- Pancreas.
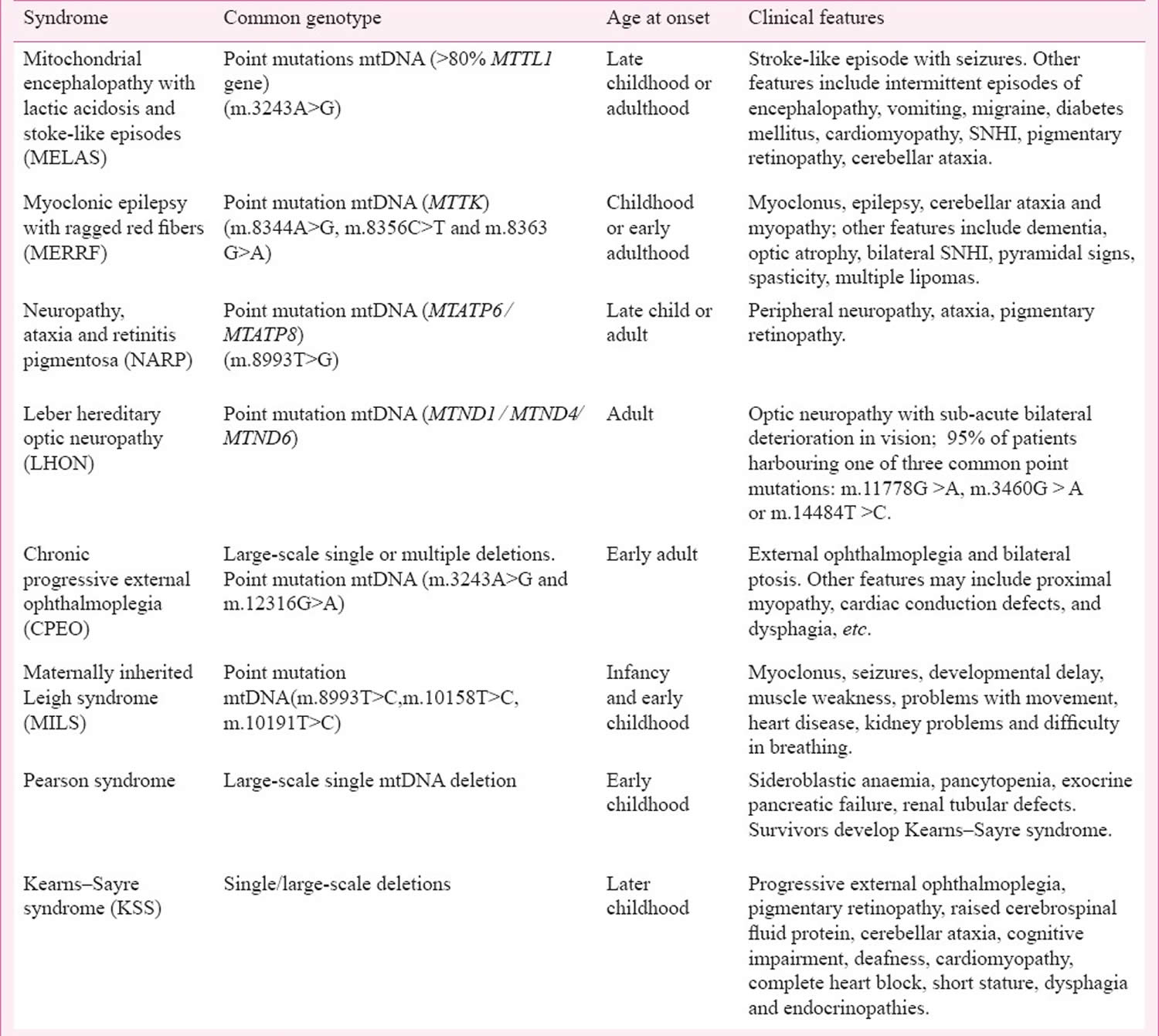
Figure 4. Mitochondrial diseases
Footnote: Clinical features and the organs affected by mitochondrial diseases.
[Source 28 ]Figure 5. Mitochondrial diseases signs and symptoms
Are mitochondrial diseases difficult to diagnose?
Yes. Because mitochondrial diseases affect so many different organs and tissues of your body, and you may have many different symptoms, mitochondrial diseases can be difficult to diagnose. There’s no single laboratory test that can diagnose a mitochondrial disease. This is why a referral to a medical facility with physicians who specialize in these diseases is critical to making the diagnosis.
CPEO disease cause
CPEO or chronic progressive external ophthalmoplegia can result from mutations in one of several different genes. In some CPEO cases, mutations occur in the POLG, TWNK, RRM2B, and SLC25A4 genes 20, 11. These genes (i.e., POLG, TWNK, RRM2B, and SLC25A4) are critical for the production and maintenance of mitochondrial DNA (mtDNA). Less commonly, mutations that change single nucleotides in genes found in mitochondrial DNA (mtDNA), such as the MT-TL1 gene, cause chronic progressive external ophthalmoplegia (CPEO). These mutations occur in genes that provide instructions for making molecules called transfer RNAs. Transfer RNAs help assemble protein building blocks (amino acids) into functioning proteins. The transfer RNAs associated with CPEO (chronic progressive external ophthalmoplegia) are present in mitochondria and help assemble the proteins that carry out the steps of oxidative phosphorylation.
There are at least 67 POLG gene mutations that cause chronic progressive external ophthalmoplegia (CPEO). Gene that has been associated with CPEO include 21, 22:
- POLG
- POLG2
- TK2
- OPA1
- RRM2B
- TWNK
- SLC25A
- RRM1
- TOP3A
- C1QBP
- DNA2
- C10ORF2
- DGUOK
- MPV17
- MGME1
- SPG7
- AFG3L2
- RNASEH1
- GMPR
Most POLG gene mutations change single amino acids in the alpha subunit of polymerase gamma (pol γ), which decreases the efficiency of mitochondrial DNA (mtDNA) replication. As in another POLG-related disorder, the most common POLG gene mutation in CPEO (chronic progressive external ophthalmoplegia) is Ala467Thr (amino acid alanine is replaced with the amino acid threonine at position 467). Ala467Thr mutation blocks the ability of the alpha subunit to attach to the beta subunits and reduces polymerase gamma (pol γ) ability to synthesize DNA. Although the mechanism is unknown, up to 60% cases of mitochondrial CPEO are due to mitochondrial DNA (mtDNA) deletions or have fewer copies of mtDNA (mtDNA depletion) 11. This abnormality (mtDNA depletion) is seen only in the tissues affected by the disease. MtDNA depletion leads to impaired oxidative phosphorylation and a decrease in cellular energy. Researchers have not determined how deletions of mtDNA lead to the specific signs and symptoms of chronic progressive external ophthalmoplegia (CPEO), although the features of the condition are probably related to impaired oxidative phosphorylation 23. It has been suggested that eye muscles are commonly affected by mitochondrial defects because they are especially dependent on oxidative phosphorylation for energy.
Enzymes encoded by mitochondrial DNA (mtDNA) play a key role in oxidative phosphorylation necessary to meet the metabolic demands of active muscle 24. To maintain a high fatigue resistance, eye muscles that control eye movement (extraocular muscles) have adapted to have both a higher mitochondrial content and higher metabolic demand compared to skeletal muscles (the most common type of muscle in your body which is connected to your bones to form part of the skeletal system which moves your limbs and other parts of your body). These properties are hypothesized to be one of the reasons why eye muscles that control eye movement (extraocular muscles) are especially vulnerable to the oxidative phosphorylation dysfunction that occurs in CPEO (chronic progressive external ophthalmoplegia) 24.
Table 1. Gene mutations that cause mitochondrial progressive external ophthalmoplegia
| Gene | Protein | PEO Phenotype | Inheritance |
|---|---|---|---|
| TYMP | Thymidine phosphorylase | MNGIE | Autosomal recessive |
| SLC25A4 | Adenine nucleotide translocator 1 | PEO | Autosomal dominant or autosomal recessive |
| TK2 | Thymidine kinase 2 | PEO/PEO-plus | Autosomal recessive |
| DGUOK | Deoxyguanosine kinase | PEO | Autosomal recessive |
| POLG | Polymerase gamma catalytic subunit | PEO/PEO-plus/SANDO/MNGIE-like | Autosomal dominant or autosomal recessive |
| TWNK | Twinkle | PEO | Autosomal dominant or autosomal recessive |
| POLG2 | Polymerase gamma subunit 2 | PEO | Autosomal dominant or autosomal recessive |
| RRM2B | P53-subunit of ribonucleotide reductase | PEO/KSS/MNGIE-like | Autosomal dominant or autosomal recessive |
| OPA1 | GTPase mitochondrial fusion | PEO-plus | Autosomal dominant |
| MGME1 | Mitochondrial genome maintenance exonuclease 1 | PEO/KSS | Autosomal recessive |
| DNA2 | DNA replication ATP-dependent | PEO | Autosomal dominant |
| SPG7 | Paraplegin | PEO-plus | Autosomal recessive |
| AFGL2 | AFG3-like protein 2 | PEO-plus | Autosomal dominant |
| RNASEH1 | Ribonuclease H1 | PEO/PEO-plus | Autosomal recessive |
| C1QBP | Complement component C1q binding protein | PEO | Autosomal recessive |
| TOP3A | DNA topoisomerase 3 alpha | PEO-plus | Autosomal recessive |
| GMPR | GMP reductase 1 | PEO | Autosomal dominant |
| LIG3 | Ligase III | MNGIE-like | Autosomal recessive |
| RRM1 | Ribonucleotide reductase catalytic subunit | PEO/MNGIE-like | Autosomal dominant or autosomal recessive |
Abbreviations: KSS = Kearns–Sayre syndrome; MNGIE = mitochondrial neurogastrointestinal encephalomyopathy; PEO = progressive external ophthalmoplegia; SANDO = sensory ataxic neuropathy dysarthria ophthalmoplegia.
[Source 2 ]CPEO inheritance pattern
CPEO (chronic progressive external ophthalmoplegia) can have different inheritance patterns depending on the gene involved.
- When the nuclear genes POLG, TWNK, RRM2B, or SLC25A4 are involved, CPEO (chronic progressive external ophthalmoplegia) is usually inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the disorder. This CPEO (chronic progressive external ophthalmoplegia) is better known as Autosomal Recessive Progressive External Ophthalmoplegia (arPEO).
- Certain mutations in the POLG or RRM2B gene can also cause a form of CPEO (chronic progressive external ophthalmoplegia) that is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. This CPEO (chronic progressive external ophthalmoplegia) is better known as Autosomal Dominant Progressive External Ophthalmoplegia (adPEO).
- When CPEO (chronic progressive external ophthalmoplegia) is caused by mutations in the MT-TL1 gene and other mitochondrial transfer RNA genes, it is inherited in a mitochondrial pattern, which is also known as maternal inheritance. This pattern of inheritance applies to genes contained in mtDNA. Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can inherit disorders resulting from mtDNA mutations only from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
- Single, large deletions of mtDNA are typically not inherited but occur during the formation of a mother’s egg cells or in early development of the embryo. Sporadic cases of CPEO suggest de novo mutations in mtDNA also called new mutation, which is a genetic alteration that arises spontaneously in an individual and is not inherited from either parent. These mutations can occur in germ cells (sperm or egg) or in the fertilized egg during early embryonic development. Individuals with these mutations usually have no history of the disorder in their family.
Because CPEO (chronic progressive external ophthalmoplegia) is a inherited genetic disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Autosomal Recessive Progressive External Ophthalmoplegia (arPEO)
Progressive CPEO without systemic involvement is the hallmark of Autosomal Recessive Progressive External Ophthalmoplegia (arPEO). Caution needs to be exercised, however, when making the diagnosis of Autosomal Recessive Progressive External Ophthalmoplegia (arPEO), as some POLG genetic mutations associated with Autosomal Recessive Progressive External Ophthalmoplegia (arPEO) are also associated with ataxia neuropathy spectrum (ANS) and other POLG-related disorders with systemic involvement. Therefore, many individuals who have no other clinical findings at the time of diagnosis with isolated Autosomal Recessive Progressive External Ophthalmoplegia (arPEO) develop other signs and symptoms of POLG-related disorders over subsequent years or decades 29, 30, 31.
Autosomal Dominant Progressive External Ophthalmoplegia (adPEO)
The universal signs and symptoms of this adult-onset progressive external ophthalmoplegia (CPEO) is progressive weakness of the extraocular eye muscles resulting in ptosis and strabismus 29. A generalized myopathy is present in most affected individuals, leading to early fatigue and exercise intolerance. Some affected individuals have variable degrees of sensorineural hearing loss, axonal neuropathy, ataxia, depression, parkinsonism, hypogonadism, and cataracts 32, 33. Cardiomyopathy (heart muscle disease) and gastrointestinal dysmotility are less common.
CPEO disease signs and symptoms
CPEO signs and symptom may include the following:
- Drooping eyelids (ptosis)
- Paralysis of the muscles that control eye movement (ophthalmoplegia)
- Impaired swallowing (dysphagia)
- Weakness of the limbs (myopathy).
- CPEO may also be associated with hearing loss, loss of sensation in the limbs, impaired muscle coordination, movement abnormalities, cardiac conduction block, or depression. In these cases, the disorder is referred to as chronic progressive external ophthalmoplegia plus (CPEO+)34.
CPEO most commonly begins in the third or fourth decade of life 18, 11. Patients with CPEO typically present with bilateral (both eyes), symmetric, painless, and pupil-spared ptosis (drooping eyelids) and weakness or paralysis of the muscles that move the eye (ophthalmoplegia). As the name implies, CPEO takes a slowly progressive course, over a course of years, making it distinct from other diseases or conditions that cause acute/subacute or static forms of weakness or paralysis of the muscles that move the eye (ophthalmoplegia) 11, 35, 18. With bilateral (both eyes) and symmetrical involvement of the eyes, patients can be asymptomatic without double vision (diplopia) even with severe defects in eye movement 11. They will also compensate for their lack of eye movement by moving their head 36. In addition, due to the gradual course, patients may not notice the eyelid droop unless it is brought up by a another person 18. Other less common eye signs include pigmentary retinopathy and optic atrophy 11, 18, 36, 37. However, there are cases that may present without ptosis or one eye affected (asymmetric).
Pain, proptosis, periorbital swelling, lid lag or retraction and pupil involvement are not symptoms of CPEO and indicate a different cause 10). Unilateral (one eye involvement) or rapidly progressive symptoms are also atypical and should prompt additional evaluation including neuroimaging 18. Detailed family history is important in identifying possible inherited conditions.
Other non-eye signs and symptoms include sensorineural hearing loss and difficulty swallowing (dysphagia) 38.
Although muscle weakness is the primary symptom of CPEO (chronic progressive external ophthalmoplegia), this condition can be accompanied by other signs and symptoms. In these instances, the condition is referred to as chronic progressive external ophthalmoplegia plus (CPEO+). Additional signs and symptoms can include hearing loss caused by nerve damage in the inner ear (sensorineural hearing loss), weakness and loss of sensation in the limbs due to nerve damage (neuropathy), impaired muscle coordination (ataxia), a pattern of movement abnormalities known as parkinsonism, and depression.
Kearns–Sayre Syndrome
Kearns–Sayre syndrome (KSS) is a subtype of CPEO with bilateral pigmentary retinopathy, a degenerative condition of the retina that impairs function of pigment epithelial cells and photoreceptors. Unlike isolated CPEO that commonly presents at the third or fourth decade of life, patients with Kearns–Sayre syndrome typically have symptom onset before age 20 as well as one of the following features: a complete heart block, cerebellar ataxia, dementia, deafness, short stature, endocrine abnormalities, and cerebrospinal fluid (CSF) protein of more than 100 mg/dL 39. If the diagnostic criteria are not met, the patient is termed “CPEO plus” (CPEO+) or “KSS-minus” 40.
Kearns–Sayre syndrome patients experience a progressive loss of peripheral and night vision. Therefore, a dilated fundus exam should be performed in cases of CPEO to visualize any areas of hypo- or hyper-pigmentation indicating retinal pigment epithelium (RPE) disturbances that might suggest Kearns–Sayre syndrome 18. The atypical retinal pigment epithelium (RPE) depigmentation has been described as having a “salt-and-pepper” or “moth-eaten” appearance 11. Other associated findings in Kearns–Sayre syndrome (KSS) include cardiovascular conduction defects, elevated protein in the cerebrospinal fluid (CSF), and cerebellar ataxia. Patients with Kearns–Sayre syndrome (KSS) should therefore be considered for cardiac assessment, a lumbar puncture, and a thorough neurologic assessment 11, 39.
When a patient presents with CPEO before the age of 20, they should be evaluated with mtDNA genetic testing followed by regular ophthalmologic assessments with dilated fundus exam and screening for systemic signs and symptoms. A muscle biopsy can be performed to look for the ragged red fibers (RRF). The fundoscopic examination reveals pigmentary retinopathy that should be distinguished from retinitis pigmentosa since they might share similar symptoms like mildly reduced night vision and visual acuity. Retinitis pigmentosa (RP) is a group of inherited eye disorders that cause damage to the retina, the light-sensitive tissue at the back of the eye. This damage leads to a progressive loss of vision, starting with night vision and peripheral (side) vision. Retinitis pigmentosa typically affects the peripheral or the mid-peripheral retina with a bone spicule pattern, whereas Kearns–Sayre syndrome affects the posterior retina with a salt and pepper pattern 41.
It is essential to perform an electrocardiogram (ECG) on Kearns–Sayre syndrome patients to rule out a complete heart block. Endocrine abnormalities affecting the adrenals, parathyroid, and hypothalamus can present with diabetes mellitus, growth hormone deficiency, and short stature 42, 43. Orbicularis oculi muscle weakness can impair eyelid closure, and frontalis weakness can affect eyelid elevation. Difficulty swallowing (dysphagia) is a rare presentation of Kearns–Sayre syndrome and may result from upper esophageal sphincter dysfunction and reduced peristalsis in the pharynx and upper esophagus, as observed in a manometric study of a case report 44.
No definitive treatment option is available for Kearns–Sayre syndrome. Symptomatic treatment includes correction of CPEO, treating heart blocks with pacemakers with a long-term cardiology follow-up, correction of endocrine abnormalities, and cochlear implants in cases of hearing loss.
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE)
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a rare, autosomal recessive disorder characterized by a progressive multisystem disorder that primarily affects the gastrointestinal tract (progressive gastrointestinal dysmotility manifesting as early satiety, nausea, dysphagia, gastroesophageal reflux, postprandial emesis, episodic abdominal pain and/or distention, and diarrhea and cachexia), nervous system (demyelinating peripheral neuropathy and leukoencephalopathy manifesting as paresthesias [tingling, numbness, and pain] and symmetric and distal weakness more prominently affecting the lower extremities), and progressive external ophthalmoplegia (PEO) 45. The order in which signs and symptoms appear is unpredictable 45. Onset is usually between the first and fifth decades; in about 60% of individuals, symptoms begin before age 20 years with a mean age of onset around 18 years 45.
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is caused by mutations in the TYMP gene, which codes for thymidine phosphorylase (TP) enzyme, resulting in loss of thymidine phosphorylase (TP) activity leading to accumulation of thymidine and deoxyuridine in the blood and tissues and mitochondrial deoxynucleoside triphosphate pool imbalances that cause mtDNA instability 46, 47, 45.
Patients with MNGIE or MNGIE-like signs and symptoms have also been linked to POLG, RRM2B, and LIG3 gene mutations 48, 49, 50 as well as mtDNA point mutations 51, 52.
Laboratory tests indicate mitochondrial dysfunction with elevated lactate in blood or cerebrospinal fluid (CSF), or both; mtDNA multiple deletions, or somatic point mutations; and ragged-red fibers (RRFs), COX-deficient muscle fibers, or both in muscle 53.
Typically, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) onset is in the late teens and progresses to fatality in the late-30s 54. Allogeneic hematopoietic stem cell transplantation (allo-HSCT that involves replacing a patient’s damaged bone marrow with healthy stem cells from a donor) and liver transplantation decrease levels of the toxic nucleosides, thymidine and deoxyuridine, and may lead to stabilization or mild clinical improvements; however, due to high morbidity and mortality after allo-HSCT, liver transplantation is the more suitable option for most MNGIE patients 55, 56, 53.
Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO)
Sensory Ataxic Neuropathy Dysarthria Ophthalmoplegia (SANDO) is a very rare mitochondrial disease that is part of the ataxia neuropathy spectrum (ANS) typically associated with autosomal recessive mutations in the POLG1 gene 57, 58. Rare cases of SANDO have been identified to be caused by mutations in C10orf2 and RNASEH1 genes 59, 60, 61. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) primarily affects the brain, muscles, nerves, and eyes 62, 8. Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) usually presents during adulthood; the mean age of onset is 32 years, although it can also be found in younger patients 63. The symptoms caused by sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) and their severity vary widely among individuals with the disorder. While the disorder worsens over time, its progression is slower and milder than other related mitochondrial disorders 8.There is no cure for SANDO. Available treatments focus on symptom management and improving an individual’s quality of life.
Pearson syndrome
Pearson Syndrome also known as Pearson marrow-pancreas syndrome, is a rare fatal multisystemic mitochondrial disorder due to deletions in mitochondrial DNA (mtDNA) that is characterized by bone marrow failure and pancreatic dysfunction 64. Pearson syndrome typically presents in infancy with anemia and can lead to failure to thrive and other multisystem complications. Pearson syndrome is caused by single, large deletions of mitochondrial DNA (mtDNA), which can range from 1,000 to 10,000 DNA building blocks (nucleotides) 64. The most common deletion, which occurs in about 20 percent of affected individuals, removes 4,997 nucleotides 64. The mtDNA deletions involved in Pearson syndrome result in the loss of genes that provide instructions for proteins involved in oxidative phosphorylation 64. These deletions impair oxidative phosphorylation and decrease the energy available to cells. It is not clear how loss of mtDNA leads to the specific signs and symptoms of Pearson syndrome, although the features of Pearson syndrome are likely related to a lack of cellular energy. Pearson syndrome causes problems with the development of blood-forming (hematopoietic) cells in the bone marrow that have the potential to develop into different types of blood cells. For this reason, Pearson syndrome is considered a bone marrow failure disorder. Function of the pancreas and other organs can also be affected. Some people with Pearson syndrome have signs and symptoms that include droopy eyelids (ptosis), CPEO, corneal endothelial dysfunction, mild peripheral pigmentary retinopathy, vision problems, hearing loss, seizures, or movement disorders 65.
About half of children with Pearson syndrome die in infancy or early childhood. Usually, premature death at three years of age occurs due to infection from neutropenia, severe lactic acidosis or liver failure. Many of those who survive develop signs and symptoms later in life of a related disorder called Kearns-Sayre syndrome. Kearns-Sayre syndrome causes weakness of muscles around the eyes and other problems. Therefore, early diagnosis is essential in improving the poor prognosis for these patients. The diagnosis of Pearson syndrome is challenging due to the atypical presentation in infancy. It can be confirmed via mitochondrial DNA (mtDNA) sequencing and observing multiple deletions of varying lengths 66. Interestingly, these single large-scale mitochondrial DNA deletions (mtDNA deletions) can also be found in young patients with CPEO and Kearns–Sayre syndrome. Together they form a continuous spectrum of diseases termed “mtDNA deletion syndromes”, supported by reports of a Kearns–Sayre syndrome-like phenotype in Pearson syndrome survivors 67.
Most affected individuals have a shortage of red blood cells (anemia), which can cause pale skin (pallor), weakness, and fatigue. Some of these individuals also have low numbers of white blood cells (neutropenia) and platelets (thrombocytopenia). Neutropenia can lead to frequent infections; thrombocytopenia sometimes causes easy bruising and bleeding. When visualized under the microscope, bone marrow cells from affected individuals may appear abnormal. Often, early blood cells (hematopoietic precursors) have multiple fluid-filled pockets called vacuoles. In addition, red blood cells in the bone marrow can have an abnormal buildup of iron that appears as a ring of blue staining in the cell after treatment with certain dyes. These abnormal cells are called ring sideroblasts and the anemia is called sideroblastic anemia.
In people with Pearson syndrome, the pancreas does not work as well as usual. The pancreas produces and releases enzymes that aid in the digestion of fats and proteins. Reduced function of this organ can lead to high levels of fats in the liver (liver steatosis). The pancreas also releases insulin, which helps maintain correct levels of blood glucose, also called blood sugar. A small number of individuals with Pearson syndrome develop diabetes mellitus, a condition characterized by abnormally high blood glucose levels that can be caused by a shortage of insulin. In addition, affected individuals may have scarring (fibrosis) in the pancreas.
Due to exocrine pancreatic dysfunction, people with Pearson syndrome have a reduced ability to absorb nutrients from the diet (malabsorption), and most affected infants have an inability to grow and gain weight at the expected rate (failure to thrive). Another common occurrence in people with Pearson syndrome is buildup in the body of a chemical called lactic acid (lactic acidosis), which can be life-threatening. In addition, liver and kidney problems can develop in people with Pearson syndrome.
Pearson syndrome can also present with pancytopenia, failure to thrive, diarrhea, hypospadias, cleft lip palate, renal tubular dysfunction, hepatic failure, enteropathy, and rashes 68. Heart conditions such as bundle branch blocks and supraventricular tachycardia, have been reported; however, heart involvement is not yet a part of the major criterion of Pearson syndrome 69.
Treatment for Pearson syndrome is supportive and may include blood transfusions, iron chelating therapy, pancreatic replacement therapy, and prompt detection and management of heart dysfunction. Bone marrow transplant has been tested and, unfortunately, yielded poor outcomes 68, 70.
Leigh syndrome
Leigh syndrome is a inherited severe progressive neurodegenerative disorder that affects the central nervous system (brain and spinal cord), leading to a progressive degeneration of brain tissue that is characterized by progressive loss of mental and movement abilities (psychomotor regression) and resulting in early death within two to three years, often due to respiratory failure 71, 72. Leigh syndrome is the most common infantile form of mitochondrial disease, usually becomes apparent in infants and young children of 3 months to 2 years of age and affecting around 1 in 40,000 individuals 71, 72. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly.
The first signs of Leigh syndrome seen in infancy are usually vomiting, diarrhea, and difficulty swallowing (dysphagia), which disrupts eating 72. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive). Severe muscle and movement problems are common in Leigh syndrome. Affected individuals may develop weak muscle tone (hypotonia), involuntary muscle contractions (dystonia), and problems with movement and balance (ataxia). Loss of sensation and weakness in the limbs (peripheral neuropathy), common in people with Leigh syndrome, may also make movement difficult 72. According to a meta-analysis by Chang et al. 73, the most common clinical features of Leigh syndrome were developmental delay, hypotonia, respiratory dysfunction, epilepsy, reduced feeding, and weakness.
Several other features may occur in people with Leigh syndrome. Many individuals with Leigh syndrome develop weakness or paralysis of the muscles that move the eyes (ophthalmoplegia); rapid, involuntary eye movements (nystagmus); strabismus or “lazy eyes” (a condition where the eyes do not point in the same direction with one or both eyes either turning inward [esotropia], outward [exotropia], upward [hypertropia], or downward [hypotropia]); or degeneration of the nerves that carry information from the eyes to the brain (optic atrophy) and pigmentary retinopathy 73, 74, 72. Severe breathing problems are common, and these problems can worsen until they cause acute respiratory failure 72. Some affected individuals also develop heart abnormalities such as hypertrophic cardiomyopathy (a thickening of the heart muscle that forces the heart to work harder to pump blood) or dilated cardiomyopathy (a heart muscle disease where the heart’s main pumping chamber, left ventricle, enlarges and weakens, making it difficult for the heart to pump blood efficiently) and conduction defects such as Wolff–Parkinson–White syndrome 75, 76, 72. In addition, a substance called lactate can build up in the body, and excessive amounts are often found in the blood, urine, or the fluid that surrounds and protects the brain and spinal cord (cerebrospinal fluid [CSF]) of people with Leigh syndrome 72.
The signs and symptoms of Leigh syndrome are caused in part by patches of damaged tissue (lesions) that develop in the brains of people with Leigh syndrome. A medical procedure called magnetic resonance imaging (MRI) reveals characteristic lesions in certain regions of the brain. These regions include the basal ganglia, which help control movement; the cerebellum, which controls the ability to balance and coordinates movement; and the brainstem, which connects the brain to the spinal cord and controls functions such as swallowing and breathing. The brain lesions are often accompanied by loss of the myelin coating around nerves (demyelination), which reduces the ability of the nerves to activate muscles used for movement or relay sensory information from the rest of the body back to the brain.
Leigh syndrome can be caused by multiple mitochondrial DNA (mtDNA) deletions as well as nuclear DNA (nDNA) defects in more than 110 different genes, most commonly by the SURF1 mutation 77, 78. In humans, most genes are found in DNA in the cell’s nucleus, called nuclear DNA (nDNA). However, some genes are found in DNA in specialized structures in the cell called mitochondria. This type of DNA is known as mitochondrial DNA (mtDNA). While most people with Leigh syndrome have a mutation in nuclear DNA (nDNA), about 20 percent have multiple mitochondrial DNA (mtDNA) deletions.
Most genes associated with Leigh syndrome are involved in the process of energy production in mitochondria. Mitochondria use oxygen to convert the energy from food into a form cells can use through a process called oxidative phosphorylation. Five protein complexes, made up of several proteins each, are involved in this process. The complexes are named complex I, complex II, complex III, complex IV, and complex V. During oxidative phosphorylation, the protein complexes drive the production of adenosine triphosphate (ATP), the cell’s main energy source, through a step-by-step transfer of negatively charged particles called electrons. Many of the gene variants associated with Leigh syndrome affect proteins in these complexes or disrupt their assembly. These variants reduce or eliminate the activity of one or more of these complexes, which can lead to Leigh syndrome.
Disruption of complex I, also called NADH:ubiquinone oxidoreductase, is the most common cause of Leigh syndrome, accounting for nearly one third of cases of Leigh syndrome 72. At least 25 genes involved in the formation of complex I, found in either nuclear or mitochondrial DNA, have been associated with Leigh syndrome.
Disruption of complex IV, also called cytochrome C oxidase (COX), is also a common cause of Leigh syndrome, underlying approximately 15 percent of cases. One of the most frequently altered genes in Leigh syndrome is SURF1. The SURF1 gene is found in nuclear DNA (nDNA) provides instructions for making a protein that helps assemble the cytochrome C oxidase (COX) protein complex (complex IV). This complex, which is involved in the last step of electron transfer in oxidative phosphorylation, provides the energy that will be used in the next step of the process to generate ATP (adenosine triphosphate). Mutations in the SURF1 gene typically lead to an abnormally short SURF1 protein that is broken down in cells, resulting in the absence of functional SURF1 protein. The loss of this protein reduces the formation of normal cytochrome C oxidase (COX) complexes, which impairs mitochondrial energy production.
The most common mitochondrial DNA (mtDNA) mutation in Leigh syndrome affects the MT-ATP6 gene, which provides instructions for making a piece of complex V, also known as the ATP synthase protein complex. Using the energy provided by the other protein complexes, the ATP synthase complex generates ATP. MT-ATP6 gene mutations, found in approximately 10 percent of people with Leigh syndrome, block the generation of ATP. Other mitochondrial DNA (mtDNA) mutations associated with Leigh syndrome decrease the activity of other oxidative phosphorylation protein complexes or lead to reduced formation of mitochondrial proteins, all of which impair mitochondrial energy production.
Other gene mutations associated with Leigh syndrome decrease the activity of one or more oxidative phosphorylation protein complexes or affect additional steps related to energy production. For example, Leigh syndrome can be caused by variants in genes that form the pyruvate dehydrogenase complex or coenzyme Q10, both of which are involved in mitochondrial energy production. Mutations in genes that direct the replication of mtDNA or the production of mitochondrial proteins can also disrupt mitochondrial energy production.
Although the exact mechanism is unclear, researchers believe that impaired oxidative phosphorylation can lead to cell death because of decreased energy available in the cell. Certain tissues that require large amounts of energy, such as the brain, muscles, and heart, seem especially sensitive to decreases in cellular energy. Cell death in the brain likely causes the characteristic lesions seen in Leigh syndrome, which contribute to the signs and symptoms of Leigh syndrome. Cell death in other sensitive tissues may also contribute to the features of Leigh syndrome.
Consensus on the clinical diagnosis is yet to be determined; however, Leigh syndrome is suspected through the hallmarks of the disease along with findings suggestive of brainstem dysfunction in addition to T2 weighted brain MRI lesions and accessory laboratory findings 73. Brain MRI findings typically show bilateral symmetrical supra-tentorial (basal ganglia, thalamus, and sub-thalamus) and/or infra-tentorial (brainstem and dentate nuclei) lesions. A study by Ardissone et al 79 presented a predominating basal ganglia involvement of 90.2%. They also showed that both supra and infra-tentorial involvement is dominant in cases of both mtDNA (74%) and -nDNA (67%) variants, while isolated infra-tentorial variants are rare 79. Extensive research is being conducted to find genetic correlations with MRI findings of Leigh syndrome. For example, a retrospective cohort found significant associations between the SURF1 variant and inferior olivary nuclei lesions 80.
Abnormal laboratory findings may yield elevated blood, urine, and CSF lactate levels. Additional deficiencies may be observed in respiratory chain complexes through enzyme assays and pyruvate dehydrogenase complex 81. However, these laboratory findings are not consistently present. Therefore, confirmatory tests with genetic testings are required for a definitive diagnosis and the identification of specific gene mutations of Leigh syndrome 82.
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS)
MELAS is an acronym for Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes is a mitochondrial disease that affects many of the body’s systems, particularly the brain and nervous system (encephalo-) and muscles (myopathy) 83. MELAS also causes lactic acid to collect in your body (lactic acidosis) and may result in repeated events that are similar to strokes (stroke-like episodes) 83. MELAS signs and symptoms most often appear in childhood following a period of normal development, although they can begin at any age. Early symptoms may include muscle weakness and pain, recurrent headaches, loss of appetite, vomiting, and seizures 83. Most affected individuals experience stroke-like episodes beginning before age 40 83. These stroke-like episodes often involve temporary muscle weakness on one side of the body (hemiparesis), altered consciousness, vision abnormalities, seizures, and severe headaches resembling migraines. Repeated stroke-like episodes can progressively damage the brain, leading to vision loss, problems with movement, and a loss of intellectual function (dementia) 83. The exact incidence of MELAS is unknown. Mitochondrial diseases have an estimated incidence of 1 in 4,000 people. MELAS can affect anyone, but only women can pass it on to their children.
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidosis. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, and hormonal imbalances 83.
MELAS can result from mutations in one of several genes, including MT-ND1, MT-ND5, MT-TH, MT-TL1, and MT-TV 84, 84, 83. These genes are found in the mitochondrial DNA (mtDNA). Some of the genes related to MELAS provide instructions for making proteins involved in normal mitochondrial function. These proteins are part of a large enzyme complex in mitochondria that helps convert oxygen, fats, and simple sugars to energy. Other genes associated with MELAS provide instructions for making molecules called transfer RNAs (tRNAs), which are chemical cousins of DNA. These molecules help assemble protein building blocks called amino acids into full-length, functioning proteins within mitochondria. Mutations in a particular transfer RNA gene, MT-TL1, cause more than 80 percent of all cases of MELAS 83. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy. Researchers have not determined how changes in mitochondrial DNA (mtDNA) lead to the specific signs and symptoms of MELAS. They continue to investigate the effects of mitochondrial gene mutations in different tissues, particularly in the brain.
MELAS is inherited in a mitochondrial pattern, which is also known as maternal inheritance. Mitochondrial pattern of inheritance (maternal inheritance) applies to genes contained in mitochondrial DNA (mtDNA). Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
In most cases, people with MELAS inherit an altered mitochondrial gene from their mother. Less commonly, MELAS results from a new mutation in a mitochondrial gene and occurs in people with no family history of MELAS.
MELAS clinical presentations vary widely, MELAS signs and symptoms most often appear in childhood following a period of normal development, although they can begin at any age. MELAS signs and symptoms include stroke-like episodes, sensorineural hearing loss, and cognitive impairment associated with diffuse white matter injury. Less commonly, it can present with gastrointestinal signs and symptoms that include gastric perforation, ischemic colitis, segmental ileal paralysis, pseudo-obstruction, or megacolon. Endocrine dysfunction such as diabetes mellitus has also been reported in MELAS 85.
Eye signs and symptoms of MELAS include blindness over half the field of vision (hemianopia) and cortical blindness from stroke-like episodes, nystagmus, cataracts, CPEO, optic atrophy, salt and pepper pigmentary retinopathy, and macular degeneration 86.
The temporary stroke-like episodes of MELAS are characterized by nausea, vomiting, a migraine-like headache, encephalopathy, and focal seizures with or without neurological deficits. The exact pathogenic mechanism for stroke-like episodes is yet to be determined; however, three theories have been put forward.
- The first is insufficient energy due to mitochondrial dysfunction, supported by the increase in lactate peaks and decreased N-acetyl aspartate peaks of the occipital regions in brain magnetic resonance spectroscopy (MRS) 87.
- The second is nitric oxide (NO) deficiency, which usually regulates oxygenation and blood flow. This hypothesis is supported by a reduction in nitric oxide (NO) metabolites during acute attacks and an increase in nitric oxide (NO) synthase inhibitors in the COX-negative fibers of MELAS patients 88.
- The third theory is mitochondrial angiopathy, an accumulation of mitochondria in the smooth muscle cells and endothelial cells of small cerebral arteries leading to the narrowing of the lumen of blood vessels and reducing perfusion 89.
MRI findings of stroke-like episode exhibit stroke-like lesions that are usually differentiated from other pathologies by initially observing cortical and deep white matter lesions, in addition to occipital and parietal lobe lesions or lesions not confined to arterial territories. Perfusion-weighted imaging (PWI) or arterial spin labeling (ASL) can also show hyperperfused lesions, and magnetic resonance spectroscopy exhibits lactate peaks 90. Another distinctive finding in neuroimaging was reported in some cases of MELAS as cerebellar lesions stroke-like lesions 90, 91.
Since MELAS is associated with reduced levels of citrulline and arginine, which are nitric oxide (NO) precursors, and decreased nitric oxide (NO) that contributes to stroke-like episodes, supplement replacement with arginine was proposed. A systematic review by Argudo et al 92 concluded that the studies conducted showed promising results in managing stroke-like episodes. Acute phase management consists of giving an intravenous dose of 500 mg/kg/day or 10 g/m² in 24 hour for 3–5 days. Whereas chronically, 150–300 mg/kg/day (maximum of 500 mg) is used instead 93. A study conducted by Pek et al 84 using induced pluripotent stem cell-derived endothelial cells vouched for edaravone, a potent antioxidant, to be used for improving the vascular function in MELAS since it scavenges reactive oxygen species (ROS) and inhibits the inflammatory response in cerebrovascular diseases, which L-arginine and citrulline do not tackle. For treating epilepsy, levetiracetam is considered to be the first-line anticonvulsant in mitochondrial encephalomyopathy due to the mitochondrial toxicity of other anticonvulsant agents 94.
CPEO disease diagnosis
A referral to a medical facility with physicians who specialize in POLG diseases is critical to making the diagnosis as diagnosis of mitochondrial disorders is still a major challenge 95.
To diagnose CPEO your doctor may perform or recommend the following 8:
- Genetic testing for POLG gene mutation(s)
- Muscle biopsy for detecting mitochondrial DNA mutations. Modified-Gomori trichrome histochemical stain to skeletal muscle revealed ragged-red fibers, a sign of abnormal proliferation of mitochondria in subsarcolemmal regions of the skeletal muscle of patients with progressive ophthalmoplegia 96, 97, 98, 99.
- Heart evaluation with echocardiogram and electrocardiography (ECG) should be considered for all patients with CPEO to rule out heart changes and conduction defects 100.
- Brain-imaging such as computed tomography (CT) scan or magnetic resonance imaging (MRI) to look for changes in the brain associated with POLG disease
- Electroencephalogram (EEG or ‘brain wave’) testing for seizures and related brain issues.
Because CPEO is an inherited disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Physical exam
A comprehensive physical exam, with focus on the eye and neurological components, is crucial in identifying different types of CPEO and its associated syndromes 10), 101. The severity of the ophthalmoplegia can be quantified by measuring the uniocular fields of fixation and ductions. The Goldmann perimeter can be used to map the range of extraocular movement (EOM) in the various fields of gaze. For example, in the right eye, the cardinal axes are 0o (lateral rectus), 67o (superior rectus), 141o (inferior oblique), 180o (medical rectus), 216o (superior oblique), and 293o (inferior rectus) 10). During the eye exam, one eye is occluded and the patient follows an illuminated target along each of the axes until central fixation on the target is lost 10). In one study 102, overall mean range of extraocular movement was decreased by 73% in patients with CPEO compared to controls. The degree of ptosis can be measured by the vertical fissure height (VFH), levator function (LF), and margin-reflex distance (MRD) 101.
Laboratory studies
While CPEO is a clinical diagnosis, laboratory studies can aid in confirming the diagnosis, as well as ruling out alternate diagnoses 10). Serum lactate and creatinine kinase (CK) and CSF lactate levels may be elevated in CPEO, but this finding is neither sensitive or specific 11, 38. Absence of anti-acetylcholinesterase antibodies and thyroid autoantibodies can help in excluding for myasthenia gravis and thyroid-associated ophthalmopathy, respectively, when history alone is not sufficient 18.
Muscle histochemistry
Histological and histochemical findings are one of the most recognized and standard approaches used for the diagnosis of mitochondrial disorders. A fresh biopsy of skeletal muscle or needle biopsy is essential for the histochemical analysis of oxidative phosphorylation defect. Hematoxylin and Eosin (HE) staining is generally used for histological examination. These dyes are essential for recognizing various tissue types and the morphological changes and structural information that form the basis for many diseases (Figure 6A). Presence of “Ragged Red Fibers” (RRF) on staining with modified gomorie trichrome (MGT) is generally indicative of an abnormal subsarcolemmal accumulation of mitochondria (Figure 6B), which is one of the important features of mitochondrial disorders 103. The activity for succinate dehydrogenase (SDH) is used for identifying complex 2 disorders (Figure 6C), and is unaffected by mtDNA mutations 104. However, presence of “Ragged Red Fibers” (RRF) in other muscle diseases, and in the muscles of healthy athletes 105 poses a specific challenge in considering this as a gold standard method only for mitochondrial disorders. Presence of cytochrome C oxidase (COX) negative fibers are generally indicative of decrease in complex 4 activity (Figure 6D), and suggestive of mitochondrial dysfunction 106. Most of the patients generally show mosaic pattern of COX (cytochrome C oxidase) deficient fibers, which is due to heteroplasmic mutations and difference in the percentage of mutant mtDNA in different cells 107. Sequential reaction of cytochrome C oxidase (COX) and succinate dehydrogenase (SDH) histochemical activities is helpful, when identifying very low levels of COX negative fibers. The uniform decrease in cytochrome C oxidase (COX) activity in muscles is generally indicative of mutations in nuDNA either encoding COX subunits or COX assembly factors, such as SURF1, SCO2, etc. 108. However, in many cases the patients having high level of mitochondrial tRNA (MT-tRNA) mutations, or large scale deletions also show global decrease in COX activity 109. Caution should be taken in diagnosis of elderly patients, who generally show cytochrome C oxidase (COX) negative muscle fibers due to age related accumulation of mtDNA deletions within individual muscle fibers 110.
Although the electron microscopic changes in mitochondria, such as abnormal mitochondria (size and shape), abnormal cristae pattern and paracrystalline inclusions (Figure 6E & F) provide additional information and aid in diagnosis, but these are not considered as a standard procedure for diagnosis of mitochondrial diseases 111. Also, certain classes of mitochondrial disorders such as Leber’s hereditary optic neuropathy (LHON), neuropathy, ataxia and retinitis pigmentosa (NARP) and maternally inherited Leigh syndrome (MILS) due to mtDNA mutations, do not show abnormalities in histopathology 28. Enzyme activity for the respiratory chain complexes could be assessed in isolated mitochondria from tissue or cultured cells by spectrophotometry 28. These complexes are either studied in isolation or in linkage with other complexes and it should be taken into consideration that poorly prepared samples or other enzyme deficiencies may also lead to secondary respiratory chain deficiencies 112.
Characteristic findings on muscle biopsy can also distinguish between different underlying myopathies.
- Kearns-Sayre Syndrome (KSS) is a clinical subtype of chronic progressive external ophthalmoplegia (CPEO) 40. Kearns-Sayre Syndrome (KSS) is defined by the following triad: onset before the age of 20, CPEO, and pigmentary retinopathy 40. Affected individuals have at least 1 of the following conditions: complete heart block, cerebrospinal fluid protein greater than 100mg/dL, cerebellar ataxia, short stature, deafness, dementia, and endocrine abnormalities. Kearns-Sayre Syndrome (KSS) is primarily caused by deletions in mitochondrial DNA (mtDNA), affecting multiple organ systems. Kearns-Sayre Syndrome will show cytochrome C oxidase negative fibers with ragged red fibers on Gomori’s trichome stain 39.
- Oculopharyngeal muscular dystrophy (OPMD) is a rare, genetic, late-onset muscle disease characterized by progressive drooping eyelids (ptosis) and difficulty swallowing (dysphagia). Oculopharyngeal muscular dystrophy (OPMD) primarily affects the muscles around the eyes and throat, but can also eventually affect the limbs. Symptoms typically appear after the age of 40, with some cases presenting earlier. Oculopharyngeal muscular dystrophy (OPMD) reveal basophilic rimmed vacuoles and filamentous intranuclear inclusions 113.
- Muscle biopsies of patients with myotonic dystrophy exhibits internal nuclei, ring fibers, sarcoplasmic masses, and early type 1 fiber atrophy 114, 115.
Figure 6. Muscle histochemistry
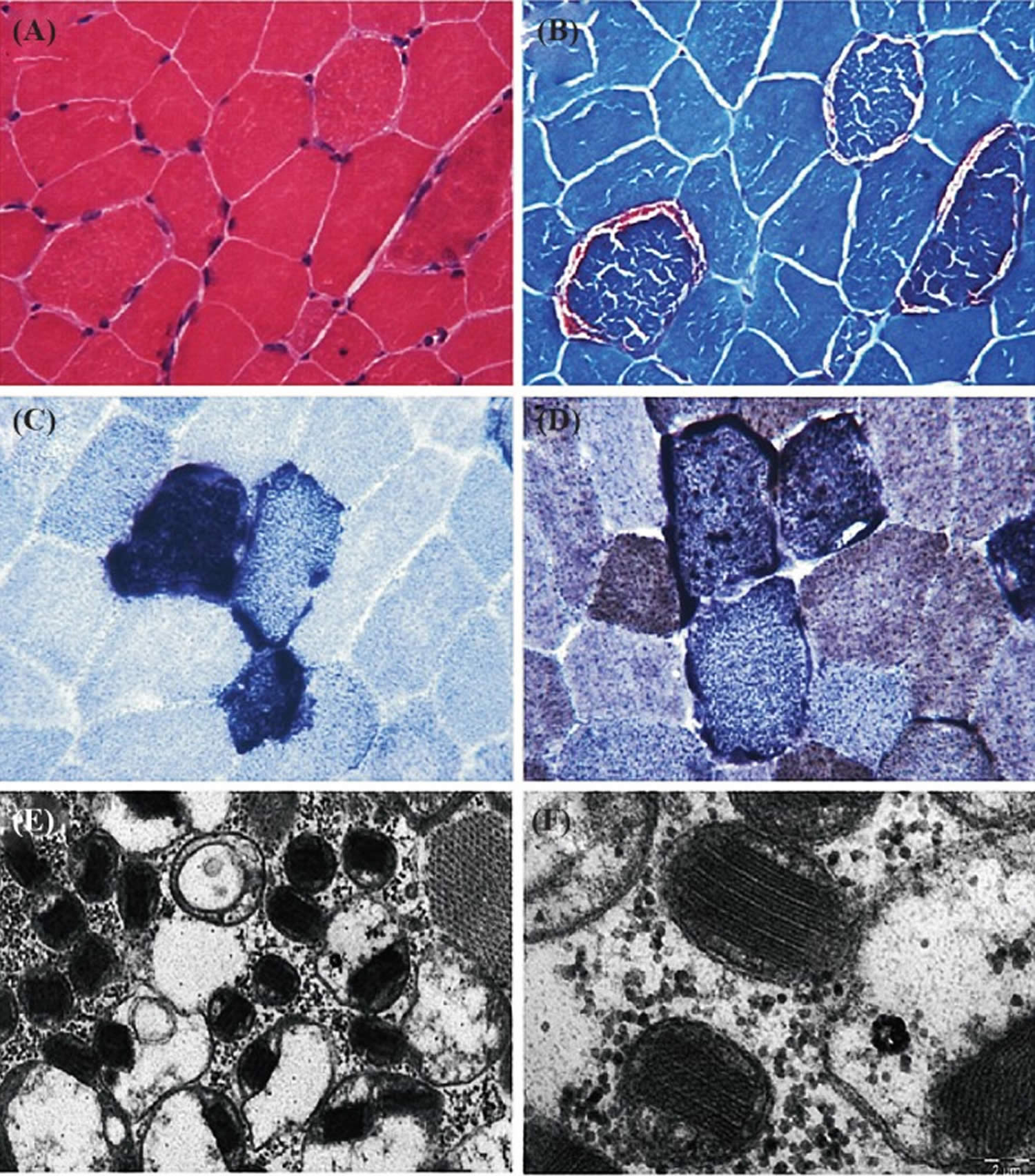
Footnotes: Tissue section showing the pathological abnormalities of mitochondrial disorders. (A) Hematoxylin and Eosin (HE) stain; (B) Red ragged fibers (RRF) appear on modified gomorie trichome stain suggesting abnormal subsarcolemmal accumulation of mitochondria; (C) Blue red fibers on succinate dehydrogenase (SDH) staining, suggesting an increased number of mitochondria; (D) COX staining shows absence of enzymes of respiratory chain. (E & F) Electron micrograph showing low and high magnification of mitochondria with paracrystalline inclusions (E x10000, F x40000).
[Source 28 ]Genetic testing
Genetic investigation for patients with suspected mitochondrial disorders should be undertaken after detailed clinical, biochemical and histopathological examination. If clinical or family history point to a specific syndrome, genetic testing may be invaluable in determining prognosis and additional studies that need to be done. Although high levels of mutations can be detected in blood, it is always advisable to use skeletal muscle for detecting mtDNA mutations, especially for deletions or mtDNA rearrangements 28. An experienced laboratory may be necessary in conducting the genetic analysis given the extensive list of potential nuclear and mitochondrial DNA involved.
Genetic testing to confirm diagnosis may include mtDNA: SLC25A4, TWNK, POLG2, SPG7, DNA2, RNASEH1, TOP3A, TK2, DGUOK, RRM2B, and GMPR
Imaging of the orbit
Orbital magnetic resonance imaging (MRI) often reveals atrophy of extraocular muscles in CPEO patients 10). One study showed a 43% decrease in the cross-sectional area of extraocular muscles in CPEO patients compared to controls 102, 116.
Brain MRI findings are numerous and nonspecific, including white matter hyperintensity, cortical atrophy, cerebellar atrophy, and brainstem hyperintensity 117.
Optic Coherence Tomography (OCT) may show reduced thickness of the outer retinal layer and macular central fovea as well as reduced volume of the optic nerve head and rim 6.
CPEO differential diagnosis
CPEO differential diagnosis include 10), 3:
- Myasthenia gravis
- Thyroid-associated ophthalmopathy (Graves’ disease)
- Multiple sclerosis (MS)
- Miller Fisher syndrome
- Congenital fibrosis of extra-ocular muscles
- Ocular sarcoidosis
- Botulism
- Cranial nerve 3 (oculomotor nerve) palsy
- Tolosa-Hunt syndrome
- Wall-Eyed Bilateral Internuclear Ophthalmoplegia (WEBINO), is a rare neurological condition characterized by bilateral adduction impairment (difficulty moving the eyes inward) and exotropia (eyes turning outward) in primary gaze 118. Wall-Eyed Bilateral Internuclear Ophthalmoplegia (WEBINO) is caused by lesions in the bilateral medial longitudinal fasciculus (MLF), a pathway in the brainstem that connects the eye movement nuclei. WEBINO is often associated with multiple sclerosis, but can also be caused by brainstem strokes or other conditions.
- CAPOS syndrome is a rare inherited neurological disorder characterized by Cerebellar ataxia, Areflexia, Pes cavus, Optic atrophy, and Sensorineural hearing loss (CAPOS) 119. CAPOS syndrome typically manifests in childhood after acute episodes, often triggered by febrile illnesses, with symptoms like ataxia, weakness, and lethargy. CAPOS syndrome is caused by a mutation in the ATP1A3, a gene that plays a role in neuronal function. The ATP1A3 gene provides instructions for making one part (the alpha-3 subunit) of a protein known as Na+/K+ ATPase or the sodium pump. This protein uses energy from a molecule called adenosine triphosphate (ATP) to transport charged atoms (ions) into and out of cells. Specifically, it pumps sodium ions (Na+) out of cells and potassium ions (K+) into cells. Na+/K+ ATPases that include the alpha-3 subunit are critical for normal function of nerve cells in the brain (neurons). The movement of sodium and potassium ions helps regulate the electrical activity of these cells and plays an important role in the signaling process that controls muscle movement. The activity of Na+/K+ ATPase also helps regulate cell size (volume). Additionally, Na+/K+ ATPase helps regulate a process called neurotransmitter reuptake. Neurotransmitters are chemical messengers that transmit signals from one neuron to another. After a neurotransmitter has had its effect, it must be removed quickly from the space between the neurons. The reuptake of neurotransmitters is carefully controlled to ensure that signals are sent and received accurately throughout the nervous system.
- CANOMAD (Chronic Ataxic Neuropathy, Ophthalmoplegia, IgM paraprotein, Cold Agglutinins, and anti-disialosyl antibodies) is a rare, chronic, immune-mediated polyneuropathy characterized by ataxia, ophthalmoplegia, and the presence of anti-disialosyl antibodies in the serum 120. CANOMAD is often associated with an IgM monoclonal gammopathy and cold agglutinins.
- Oculopharyngeal muscular dystrophy (OPMD) is a rare autosomal dominant late-onset muscle disease characterized by progressive drooping eyelids (ptosis) and difficulty swallowing (dysphagia) 121. Oculopharyngeal muscular dystrophy (OPMD) primarily affects the muscles around the eyes and throat, but can also eventually affect the limbs. Symptoms typically appear after the age of 40, with some cases presenting earlier. Oculopharyngeal muscular dystrophy (OPMD) is caused by defective polyalanine-binding protein 1 (PABPN1) gene and the defective PABPN1 gene leads to a buildup of PABPN1 proteins in the muscle cells and can cause failure of muscle regeneration through an unclear mechanism 113, 122. The PABPN1 clumps inside the muscle cells and may cause the cells to die. This leads to muscle weakness. Unlike some other CPEO-plus (CPEO+) syndromes, oculopharyngeal muscular dystrophy (OPMD) is not a mitochondrial myopathy. Oculopharyngeal muscular dystrophy (OPMD) is most prevalent among people of French-Canadian origin. Symptoms present at the fifth decade of life and in addition to CPEO, include bulbar symptoms such as dysphagia (pharyngeal muscle weakness), weakness of the orbicularis oculi muscle, and proximal limb weakness 11, 18.
- Oculopharyngodistal myopathy (OPDM) is a rare, hereditary muscle disease characterized by progressive weakness in the muscles of the eyes (ptosis, ophthalmoparesis), throat (dysphagia, dysarthria), and distal limbs leading to weakness and atrophy, often with rimmed vacuoles and intranuclear inclusions observed on muscle biopsy 123. Oculopharyngodistal myopathy (OPDM) is inherited in both autosomal dominant and autosomal recessive patterns. Oculopharyngodistal myopathy (OPDM) is caused by expansions of CGG or CCG repeats in genes like LRP12, LOC642361/NUTM2B-AS1, GIPC1, NOTCH2NLC, RILPL1, and ABCD3 124
- Limb-girdle muscular dystrophy with ophthalmoplegia is a combination of two conditions 125, 126. Limb-girdle muscular dystrophy is a group of inherited disorders characterized by muscle weakness primarily in the pelvic and shoulder girdles. Ophthalmoplegia, on the other hand, is a condition where there is paralysis of the muscles that control eye movement, leading to impaired eye movement and potentially double vision.
- Neurogenic ophthalmoplegia
- Anti-GQ1b syndrome
- Congenital cranial dysinnervation disorders: Moebius syndrome, CFEOM, Duane syndrome
- Lambert–Eaton myasthenic syndrome (LEMS)
- A-beta lipoproteinemia
- Supranuclear ophthalmoplegia
- Progressive supranuclear palsy (PSP)
- Hereditary ataxias
- Hereditary spastic paraplegia (HSP)
- Spastic paraplegia 7 (SPG7) and rarely spastic paraplegia 35 (SPG35)
- Spinocerebellar ataxia (SCA): SCA 1, 2, 3, 7, 9, 11, 28
- Medications: Statins
CPEO disease treatment
There is no cure for CPEO (chronic progressive external ophthalmoplegia), but treatment focuses on managing symptoms and improving quality of life 127, 128. Chronic progressive external ophthalmoplegia (CPEO) treatment may include physical therapy, surgery is the mainstay treatment for ptosis, and support for other affected organ systems. Referral for management of associated neurologic and heart disease is indicated.
Approximately one-third of CPEO patients experience constant or intermittent double vision (diplopia) 18. Prism lenses are effective in controlling double vision (diplopia) after a full orthoptic assessment of direction and magnitude of eye deviations. The prism compensates for the deviated eye(s) by refracting the light so that the image falls onto the macula of each eye. While some patients may want surgical correction of horizontal recti muscles, double vision (diplopia) or crossed eyes (strabismus) may reoccur over time due to the progressive nature of CPEO 18.
Clinically significant ptosis can be corrected surgically 129. If levator function remains moderate or good, advancement or resection of the levator palpebrae superioris may be considered to maximize the muscle’s elevation of the lid 10), 130, 131. However, typically levator function will progressively worsen resulting in minimal levator excursion 10). In cases of poor levator function, eyelid suspension to the frontalis muscle with autogenous or synthetic sling material should be considered 18, 132. Careful pre-operative evaluation and surgery by a trained oculoplastics surgeon is vital 10). Overcorrection of ptosis with surgery can result in incomplete or abnormal closure of the eyelids (lagophthalmos) and corneal exposure, leading to potentially blinding complications of exposure keratitis (damage to the cornea caused by prolonged exposure to the environment, often due to incomplete or inadequate eyelid closure), corneal ulcers, or eye perforation 11, 132. Some authors have used a frontalis sling with silicone rod to correct ptosis in CPEO 133.
Since disease of the cornea (keratopathy) is one of the most frequent complications of CPEO, fixing corneal exposure is key. There is a new proposed way to surgically treat CPEO with palpebral fissure transfer (PFT) with lower eyelid elevation with no spacer to successfully cover the cornea 134, 1.
Patient Perspective
Please note below is one patient’s perspective, not medical advice. You should always discuss with your eye doctor or CPEO specialist before copying the below action as it is unique to that person.
I have lived with CPEO for over 20 years. I learned early on how to manage symptoms to give myself the highest quality of life. In CPEO the eyelid does not close all the way and is weak. This results in the eyeball not getting the proper protection and in turn, this can cause visual disturbances (blurry and double vision, severe dry eyes.) The good news is that with a proper daily regimen, a person can improve and help their eyes. Before I knew and started my regiment, I had developed ulcers on my cornea. Since taking proactive measures, I have had none. The following are suggestions to help the environment of the eyes when living with CPEO:
- Tear duct plugs – help keep the moisture in the eye and an eye doctor can put them in, only takes a minute.
- Tape eyes shut each night – in the morning no more waking up with dry scratchy eyes. 1 inch 3MM surgical foam tape applied to each eye before going to sleep. Place an eye lubricant in each
- eye first. Then tear off about 3 inches from the roll of tape and secure the eyelid downwards (like you are pulling it down and into place.) The 3-inch tape should be horizontally placed.
- Daily use of eye lubricant drops.
- Restasis RX eye drops daily.
- Warm compresses on each eye when waking in the morning.
- Extra thick eye lubricant or gel eye drops when eyes are extra dry.
- Frontalis Sling surgery consult with an Oculoplastic Surgeon specializing in lid lifts to see if you are a candidate. Lifting the eyelids can be a game-changer enabling a better line of sight.
- Protective eyewear when outside, to shield from wind and sun.
CPEO disease prognosis
The prognosis for CPEO (chronic progressive external ophthalmoplegia) is generally guarded with a variable course and potential for severe complications 135. While isolated CPEO may not significantly impact life expectancy, CPEO combined with other symptoms (CPEO+) can lead to reduced life expectancy and increased morbidity 136, 137.
CPEO disease life expectancy
In many cases, isolated CPEO meaning CPEO without other associated conditions does not significantly impact life expectancy 136, 137. Individuals with CPEO (chronic progressive external ophthalmoplegia) generally have a normal life expectancy, especially when the disease is isolated and not accompanied by other complications. However, the prognosis can be more complex and affected by factors such as the severity of the disease, the presence of associated symptoms, and the involvement of other organs.
- Karimi N, Ghahvehchian H, Keyhani A, Manavishad A, Compton CJ, Clark JD, West NL, Kashkouli MB. Type and Frequency of Misdiagnosis and Time Lag to Diagnosis in Patients with Chronic Progressive External Ophthalmoplegia. J Ophthalmic Vis Res. 2024 Sep 16;19(3):334-339. doi: 10.18502/jovr.v19i3.13998[↩][↩]
- Hirano M, Pitceathly RDS. Progressive external ophthalmoplegia. Handb Clin Neurol. 2023;194:9-21. doi: 10.1016/B978-0-12-821751-1.00018-X[↩][↩]
- Ali A, Esmaeil A, Behbehani R. Mitochondrial Chronic Progressive External Ophthalmoplegia. Brain Sci. 2024 Jan 27;14(2):135. doi: 10.3390/brainsci14020135[↩][↩][↩][↩]
- Kierdaszuk B, Kaliszewska M, Rusecka J, Kosińska J, Bartnik E, Tońska K, Kamińska AM, Kostera-Pruszczyk A. Progressive External Ophthalmoplegia in Polish Patients-From Clinical Evaluation to Genetic Confirmation. Genes (Basel). 2020 Dec 31;12(1):54. doi: 10.3390/genes12010054[↩]
- Lv H, Qu Q, Liu H, Qian Q, Zheng X, Zhang Y. Clinical, neuroelectrophysiological and muscular pathological analysis of chronic progressive external ophthalmoplegia. Exp Ther Med. 2020 Aug;20(2):1770-1774. doi: 10.3892/etm.2020.8822[↩]
- Wu Y, Kang L, Wu HL, Hou Y, Wang ZX. Optical coherence tomography findings in chronic progressive external ophthalmoplegia. Chin Med J (Engl). 2019 May 20;132(10):1202-1207. doi: 10.1097/CM9.0000000000000262[↩][↩]
- Bermejo-Guerrero L, de Fuenmayor-Fernández de la Hoz CP, Serrano-Lorenzo P, Blázquez-Encinar A, Gutiérrez-Gutiérrez G, Martínez-Vicente L, Galán-Dávila L, García-García J, Arenas J, Muelas N, Hernández-Laín A, Domínguez-González C, Martín MA. Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. J Clin Med. 2021 Dec 22;11(1):22. doi: 10.3390/jcm11010022[↩]
- Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. 2010 Mar 16 [Updated 2024 Feb 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26471[↩][↩][↩][↩]
- Chinnery PF. Primary Mitochondrial Disorders Overview. 2000 Jun 8 [Updated 2021 Jul 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1224[↩]
- Chronic Progressive External Ophthalmoplegia (CPEO). https://eyewiki.org/Chronic_Progressive_External_Ophthalmoplegia_(CPEO[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Lee AG, Brazis PW. Chronic progressive external ophthalmoplegia. Curr Neurol Neurosci Rep. 2002 Sep;2(5):413-7. doi: 10.1007/s11910-002-0067-5[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Progressive external ophthalmoplegia. https://medlineplus.gov/genetics/condition/progressive-external-ophthalmoplegia[↩]
- Fraser JA, Biousse V, Newman NJ. The neuro-ophthalmology of mitochondrial disease. Surv Ophthalmol. 2010;55(4):299–334. doi: 10.1016/j.survophthal.2009.10.002[↩][↩]
- Chinnery PF. Mitochondrial disease in adults: what’s old and what’s new? EMBO Mol Med. 2015;7(12):1503–1512. doi: 10.15252/emmm.201505079[↩]
- Caballero PE, Candela MS, Alvarez CI, Tejerina AA. Chronic progressive external ophthalmoplegia: a report of 6 cases and a review of the literature. Neurologist. 2007;13(1):33–36. doi: 10.1097/01.nrl.0000252953.49721.f5[↩]
- Ramakrishnan S, Yadav R, Adwani S. Vocal cord palsy in a case of chronic progressive external ophthalmoplegia. Ann Indian Acad Neurol. 2015;18(4):481–483. doi: 10.4103/0972-2327.165463[↩]
- Bucelli RC, Lee MS, McClelland CM. Chronic Progressive External Ophthalmoplegia in the Absence of Ptosis. J Neuroophthalmol. 2016 Sep;36(3):270-4. doi: 10.1097/WNO.0000000000000384[↩]
- Yu-Wai-Man, P. Chronic Progressive External Ophthalmoplegia Secondary to Nuclear-Encoded Mitochondrial Genes. in Mitochondrial Case Studies 159–169[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Yu-Wai-Man P., Clements Al Nesbitt V., Griffiths P.G., Gorman G.S., Schaefer A.M., Turnbull D.M., Taylor R.W., McFarland R. A national epidemiological study of chronic progressive external ophthalmoplegia in the United Kingdom—Molecular genetic features and neurological burden. Investig. Ophthalmol. Vis. Sci. 2014;55:5109.[↩]
- Tarnopolsky, M. Chronic Progressive External Ophthalmoplegia (CPEO). in Mitochondrial Case Studies 49–53.[↩][↩]
- Van Goethem G, Martin JJ, Van Broeckhoven C. Progressive external ophthalmoplegia characterized by multiple deletions of mitochondrial DNA: unraveling the pathogenesis of human mitochondrial DNA instability and the initiation of a genetic classification. Neuromolecular Med. 2003;3(3):129-46. doi: 10.1385/NMM:3:3:129[↩][↩]
- López-Gómez C, Cámara Y, Hirano M, Martí R; 232nd ENMC Workshop Participants. 232nd ENMC international workshop: Recommendations for treatment of mitochondrial DNA maintenance disorders. 16 – 18 June 2017, Heemskerk, The Netherlands. Neuromuscul Disord. 2022 Jul;32(7):609-620. doi: 10.1016/j.nmd.2022.05.008[↩][↩]
- POLG gene. https://medlineplus.gov/genetics/gene/polg[↩][↩]
- Yu Wai Man CY, Chinnery PF, Griffiths PG. Extraocular muscles have fundamentally distinct properties that make them selectively vulnerable to certain disorders. Neuromuscul Disord. 2005 Jan;15(1):17-23. doi: 10.1016/j.nmd.2004.10.002[↩][↩][↩][↩]
- Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011;108:13534–9. doi: 10.1073/pnas.1109263108[↩]
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.[↩]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.[↩]
- Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res. 2015 Jan;141(1):13-26. doi: 10.4103/0971-5916.154489[↩][↩][↩][↩][↩][↩]
- Van Goethem G, Dermaut B, Löfgren A, Martin JJ, Van Broeckhoven C. Mutation of POLG is associated with progressive external ophthalmoplegia characterized by mtDNA deletions. Nat Genet. 2001 Jul;28(3):211-2. doi: 10.1038/90034[↩][↩]
- Lamantea E, Tiranti V, Bordoni A, Toscano A, Bono F, Servidei S, Papadimitriou A, Spelbrink H, Silvestri L, Casari G, Comi GP, Zeviani M. Mutations of mitochondrial DNA polymerase gammaA are a frequent cause of autosomal dominant or recessive progressive external ophthalmoplegia. Ann Neurol. 2002 Aug;52(2):211-9. doi: 10.1002/ana.10278[↩]
- Van Goethem G, Martin JJ, Dermaut B, Löfgren A, Wibail A, Ververken D, Tack P, Dehaene I, Van Zandijcke M, Moonen M, Ceuterick C, De Jonghe P, Van Broeckhoven C. Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord. 2003 Feb;13(2):133-42. doi: 10.1016/s0960-8966(02)00216-x[↩]
- Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, Oldfors A, Rautakorpi I, Peltonen L, Majamaa K, Somer H, Suomalainen A. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004 Sep 4-10;364(9437):875-82. doi: 10.1016/S0140-6736(04)16983-3[↩]
- Pagnamenta AT, Taanman JW, Wilson CJ, Anderson NE, Marotta R, Duncan AJ, Bitner-Glindzicz M, Taylor RW, Laskowski A, Thorburn DR, Rahman S. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006 Oct;21(10):2467-73. doi: 10.1093/humrep/del076[↩]
- Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmophegia, and complete heart block: unusual syndrome with histologic study in one of two cases. AMA Arch Ophthalmol. 1958 Aug;60(2):280-9.[↩]
- Ahmad SS, Ghani SA. Kearns-Sayre syndrome: An unusual ophthalmic presentation. Oman J Ophthalmol. 2012 May;5(2):115-7. doi: 10.4103/0974-620X.99377[↩]
- Tarnopolsky, M. Chronic Progressive External Ophthalmoplegia (CPEO). in Mitochondrial Case Studies 49–53[↩][↩]
- DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013 Aug;9(8):429-44. doi: 10.1038/nrneurol.2013.126[↩]
- Heighton JN, Brady LI, Newman MC, Tarnopolsky MA. Clinical and demographic features of chronic progressive external ophthalmoplegia in a large adult-onset cohort. Mitochondrion. 2019 Jan;44:15-19. doi: 10.1016/j.mito.2017.12.006[↩][↩]
- Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, Nakase H, Bonilla E, Werneck LC, Servidei S, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. 1989 May 18;320(20):1293-9. doi: 10.1056/NEJM198905183202001[↩][↩][↩]
- Shemesh A, Margolin E. Kearns-Sayre Syndrome. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482341[↩][↩][↩]
- Ortiz A., Arias J., Cárdenas P., Villamil J., Peralta M., Escaf L.C., Ortiz J. Macular findings in Spectral Domain Optical Coherence Tomography and OCT Angiography in a patient with Kearns–Sayre syndrome. Int. J. Retin. Vitr. 2017;3:24. doi: 10.1186/s40942-017-0077-8[↩]
- Kang Y.X., Wang Y.J., Zhang Q., Pang X.H., Gu W. A case of hypopituitarism accompanying Kearns–Sayre syndrome treated with human chorionic gonadotropin: A case report and literature review. Andrologia. 2017;49:e12711. doi: 10.1111/and.12711[↩]
- Ng Y.S., Lim A.Z., Panagiotou G., Turnbull D.M., Walker M. Endocrine Manifestations and New Developments in Mitochondrial Disease. Endocr. Rev. 2021;43:583–609. doi: 10.1210/endrev/bnab036[↩]
- Katsanos K.H., Nastos D., Noussias V., Christodoulou D., Kappas A., Tsianos E.V. Manometric study in Kearns–Sayre syndrome. Dis. Esophagus. 2001;14:63–66. doi: 10.1111/j.1442-2050.2001.00152.x[↩]
- Hirano M. Mitochondrial Neurogastrointestinal Encephalopathy Disease. 2005 Apr 22 [Updated 2016 Jan 14]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1179[↩][↩][↩][↩]
- Nishino I, Spinazzola A, Hirano M (1999). Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 283: 689–692. 10.1126/science.283.5402.689[↩]
- Spinazzola A, Marti R, Nishino I et al. (2002). Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem 277: 4128–4133. 10.1074/jbc.M111028200[↩]
- Van Goethem G, Schwartz M, Lofgren A et al. (2003c). Novel POLG mutations in progressive external ophthalmoplegia mimicking mitochondrial neurogastrointestinal encephalomyopathy. Eur J Hum Genet 11: 547–549. 10.1038/sj.ejhg.5201002[↩]
- Shaibani A, Shchelochkov OA, Zhang S et al. (2009). Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol 66: 1028–1032. 10.1001/archneurol.2009.139[↩]
- Bonora E, Chakrabarty S, Kellaris G et al. (2021). Biallelic variants in LIG3 cause a novel mitochondrial neurogastrointestinal encephalomyopathy. Brain 144: 1451–1466. 10.1093/brain/awab056[↩]
- Gamez J, Garces-Garmendia MA (2005). Paralytic ileus in a MELAS patient mimicking MNGIE. Pediatr Neurol 33: 151. 10.1016/j.pediatrneurol.2005.05.023[↩]
- Keilland E, Rupar CA, Prasad AN et al. (2016). The expanding phenotype of MELAS caused by the m.3291T > C mutation in the MT-TL1 gene. Mol Genet Metab Rep 6: 64–69. 10.1016/j.ymgmr.2016.02.003[↩]
- Hirano M, Carelli V, De Giorgio R et al. (2021). Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): position paper on diagnosis, prognosis, and treatment by the MNGIE International Network. J Inherit Metab Dis 44: 376–387. 10.1002/jimd.12300[↩][↩]
- Garone C, Tadesse S, Hirano M (2011). Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain 134: 3326–3332. 10.1093/brain/awr245[↩]
- Halter J, Schupbach W, Casali C et al. (2011). Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant 46: 330–337. 10.1038/bmt.2010.100[↩]
- D’Angelo R, Boschetti E, Amore G et al. (2020). Liver transplantation in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): clinical long-term follow-up and pathogenic implications. J Neurol 267: 3702–3710. 10.1007/s00415-020-10051-x[↩]
- Fadic R, Russell JA, Vedanarayanan VV et al. (1997). Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology 49: 239–245. 10.1212/wnl.49.1.239[↩]
- Van Goethem G, Martin JJ, Dermaut B et al. (2003a). Recessive POLG mutations presenting with sensory and ataxic neuropathy in compound heterozygote patients with progressive external ophthalmoplegia. Neuromuscul Disord 13: 133–142. 10.1016/s0960-8966(02)00216-x[↩]
- Hudson G, Deschauer M, Busse K et al. (2005). Sensory ataxic neuropathy due to a novel C10Orf2 mutation with probable germline mosaicism. Neurology 64: 371–373. 10.1212/01.WNL.0000149767.51152.83[↩]
- Hanisch F, Kornhuber M, Alston CL et al. (2015). SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J Neurol Neurosurg Psychiatry 86: 630–634. 10.1136/jnnp-2013-306748[↩]
- Bugiardini E, Poole OV, Manole A et al. (2017). Clinicopathologic and molecular spectrum of RNASEH1-related mitochondrial disease. Neurol Genet 3: e149. 10.1212/NXG.0000000000000149[↩]
- Fadic R, Russell JA, Vedanarayanan VV, Lehar M, Kuncl RW, Johns DR. Sensory ataxic neuropathy as the presenting feature of a novel mitochondrial disease. Neurology. 1997;49(1):239-245. doi:10.1212/wnl.49.1.239[↩]
- Li LX, Jiang LT, Pan YG, et al. Clinical and Molecular Features of POLG-Related Sensory Ataxic Neuropathy with Dysarthria and Ophthalmoparesis. J Mol Neurosci. 2021;71(12):2462-2467. doi:10.1007/s12031-021-01831-9[↩]
- Pearson syndrome. https://medlineplus.gov/genetics/condition/pearson-syndrome[↩][↩][↩][↩]
- Vadhul R., Halbach C.S., Areaux R.G., Jr., Berry S., Hou J.H. Endothelial dysfunction in a child with Pearson marrow-pancreas syndrome managed with Descemet stripping automated endothelial keratoplasty using a suture pull-through technique. Digit. J. Ophthalmol. 2019;25:59–64. doi: 10.5693/djo.02.2019.09.001[↩]
- Reddy J., Jose J., Prakash A., Devi S. Pearson syndrome: A rare inborn error of metabolism with bone marrow morphology providing a clue to diagnosis. Sudan. J. Paediatr. 2019;19:161–164. doi: 10.24911/SJP.106-1534158413[↩]
- Broomfield A., Sweeney M.G., Woodward C.E., Fratter C., Morris A.M., Leonard J.V., Abulhoul L., Grunewald S., Clayton P.T., Hanna M.G., et al. Paediatric single mitochondrial DNA deletion disorders: An overlapping spectrum of disease. J. Inherit. Metab. Dis. 2015;38:445–457. doi: 10.1007/s10545-014-9778-4[↩]
- Shoeleh C., Donato U.M., Galligan A., Vitko J. A Case Report on Pearson Syndrome with Emphasis on Genetic Screening in Patients Presenting with Sideroblastic Anemia and Lactic Acidosis. Cureus. 2023;15:e33963. doi: 10.7759/cureus.33963[↩][↩]
- Jennifer M.S., Cortez D. Pearson marrow-pancreas syndrome with cardiac conduction abnormality necessitating prophylactic pacemaker implantation. Ann. Noninvasive Electrocardiol. 2020;25:e12681. doi: 10.1111/anec.12681[↩]
- Faraci M., Cuzzubbo D., Micalizzi C., Lanino E., Morreale G., Dallorso S., Castagnola E., Schiaffino M.C., Bruno C., Rossi A., et al. Allogeneic bone marrow transplantation for Pearson’s syndrome. Bone Marrow Transplant. 2007;39:563–565. doi: 10.1038/sj.bmt.1705638[↩]
- Saini A.G., Chatterjee D., Bhagwat C., Vyas S., Attri S.V. Leigh syndrome in an infant: Autopsy and histopathology findings. Autops. Case Rep. 2021;11:e2021334. doi: 10.4322/acr.2021.334[↩][↩]
- Leigh syndrome. https://medlineplus.gov/genetics/condition/leigh-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Chang X., Wu Y., Zhou J., Meng H., Zhang W., Guo J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine. 2020;99:e18634. doi: 10.1097/MD.0000000000018634[↩][↩][↩]
- Han J., Lee Y.-M., Kim S.M., Han S.Y., Lee J.B. Ophthalmological manifestations in patients with Leigh syndrome. Br. J. Ophthalmol. 2015;99:528–535. doi: 10.1136/bjophthalmol-2014-305704[↩]
- Sofou K., de Coo I.F.M., Ostergaard E., Isohanni P., Naess K., De Meirleir L., Tzoulis C., Uusimaa J., Lönnqvist T., Bindoff L.A., et al. Phenotype-genotype correlations in Leigh syndrome: New insights from a multicentre study of 96 patients. J. Med. Genet. 2018;55:21–27. doi: 10.1136/jmedgenet-2017-104891[↩]
- Brecht M., Richardson M., Taranath A., Grist S., Thorburn D., Bratkovic D. Leigh Syndrome Caused by the MT-ND5 m.13513G>A Mutation: A Case Presenting with WPW-Like Conduction Defect, Cardiomyopathy, Hypertension and Hyponatraemia. JIMD Rep. 2015;19:95–100. doi: 10.1007/8904_2014_375[↩]
- Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. doi: 10.1002/ana.24551[↩]
- Kistol D., Tsygankova P., Krylova T., Bychkov I., Itkis Y., Nikolaeva E., Mikhailova S., Sumina M., Pechatnikova N., Kurbatov S., et al. Leigh Syndrome: Spectrum of Molecular Defects and Clinical Features in Russia. Int. J. Mol. Sci. 2023;24:1597. doi: 10.3390/ijms24021597[↩]
- Ardissone A., Bruno C., Diodato D., Donati A., Ghezzi D., Lamantea E., Lamperti C., Mancuso M., Martinelli D., Primiano G., et al. Clinical, imaging, biochemical and molecular features in Leigh syndrome: A study from the Italian network of mitochondrial diseases. Orphanet J. Rare Dis. 2021;16:413. doi: 10.1186/s13023-021-02029-3[↩][↩]
- Alves C.A.P.F., Teixeira S.R., Martin-Saavedra J.S., Gonçalves F.G., Russo F.L., Muraresku C., McCormick E.M., Falk M.J., Zolkipli-Cunningham Z., Ganetzky R., et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020;88:218–232. doi: 10.1002/ana.25789[↩]
- Baertling F., Rodenburg R.J., Schaper J., Smeitink J.A., Koopman W.J.H., Mayatepek E., Morava E., Distelmaier F. A guide to diagnosis and treatment of Leigh syndrome. J. Neurol. Neurosurg. Psychiatry. 2014;85:257–265. doi: 10.1136/jnnp-2012-304426[↩]
- Ogawa E., Shimura M., Fushimi T., Tajika M., Ichimoto K., Matsunaga A., Tsuruoka T., Ishige M., Fuchigami T., Yamazaki T., et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017;40:685–693. doi: 10.1007/s10545-017-0042-6[↩]
- Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. https://medlineplus.gov/genetics/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes[↩][↩][↩][↩][↩][↩][↩][↩]
- Pek N.M.Q., Phua Q.H., Ho B.X., Pang J.K.S., Hor J.-H., An O., Yang H.H., Yu Y., Fan Y., Ng S.-Y., et al. Mitochondrial 3243A>G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019;10:802. doi: 10.1038/s41419-019-2036-9[↩][↩][↩]
- Seed L.M., Dean A., Krishnakumar D., Phyu P., Horvath R., Harijan P.D. Molecular and neurological features of MELAS syndrome in paediatric patients: A case series and review of the literature. Mol. Genet. Genom. Med. 2022;10:e1955. doi: 10.1002/mgg3.1955[↩]
- Al-Enezi M., Al-Saleh H., Nasser M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 2008;15:81–86. doi: 10.4103/0974-9233.51998[↩]
- Weiduschat N., Kaufmann P., Mao X., Engelstad K.M., Hinton V., DiMauro S., De Vivo D., Shungu D. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology. 2014;82:798–805. doi: 10.1212/WNL.0000000000000169[↩]
- El-Hattab A.W., Emrick L.T., Craigen W.J., Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012;107:247–252. doi: 10.1016/j.ymgme.2012.06.018[↩]
- Yoshida T., Ouchi A., Miura D., Shimoji K., Kinjo K., Sueyoshi T., Jonosono M., Rajput V. MELAS and reversible vasoconstriction of the major cerebral arteries. Intern. Med. 2013;52:1389–1392. doi: 10.2169/internalmedicine.52.0188[↩]
- Cheng W., Zhang Y., He L. MRI Features of Stroke-Like Episodes in Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-Like Episodes. Front. Neurol. 2022;13:843386. doi: 10.3389/fneur.2022.843386[↩][↩]
- Ng Y.S., Bindoff L.A., Gorman G.S., Horvath R., Klopstock T., Mancuso M., Martikainen M.H., Mcfarland R., Nesbitt V., Pitceathly R.D.S., et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019;4:201. doi: 10.12688/wellcomeopenres.15599.1[↩]
- Argudo J.M., Moncayo O.M.A., Insuasti W., Garofalo G., Aguirre A.S., Encalada S., Villamarin J., Oña S., Tenemaza M.G., Eissa-Garcés A., et al. Arginine for the Treatment of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes: A Systematic Review. Cureus. 2022;14:e32709. doi: 10.7759/cureus.32709[↩]
- Pérez-Cruz E., González-Rivera C., Valencia-Olvera L.d.C.G. Immunonutrition for the acute treatment of MELAS syndrome. Endocrinología. Diabetes Nutr. 2022;69:144–148. doi: 10.1016/j.endien.2022.02.006[↩]
- Li J., Zhang W., Cui Z., Li Z., Jiang T., Meng H. Epilepsy Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front. Neurol. 2021;12:675816. doi: 10.3389/fneur.2021.675816[↩]
- Hikmat O, Tzoulis C, Chong WK, Chentouf L, Klingenberg C, Fratter C, Carr LJ, Prabhakar P, Kumaraguru N, Gissen P, Cross JH, Jacques TS, Taanman JW, Bindoff LA, Rahman S. The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet Med. 2017 Nov;19(11):1217-1225. doi: 10.1038/gim.2017.35. Epub 2017 Apr 27. Erratum in: Genet Med. 2019 Apr;21(4):1027. doi: 10.1038/s41436-018-0098-1[↩]
- Olson W, Engel WK, Walsh GO et al. (1972). Oculocraniosomatic neuromuscular disease with “ragged-red” fibers. Arch Neurol 26: 193–211. 10.1001/archneur.1972.00490090019001[↩]
- Rowland LP, Blake DM, Hirano M, Di Mauro S, Schon EA, Hays AP, Devivo DC. Clinical syndromes associated with ragged red fibers. Rev Neurol (Paris). 1991;147(6-7):467-73.[↩]
- Karimi N, Kashkouli MB. Reply to the letter “Long-Term Results of Palpebral Fissure Transfer With No Lower Eyelid Spacer in Chronic Progressive External Ophthalmoplegia”. Am J Ophthalmol. 2022;236:320. doi: 10.1016/j.ajo.2021.11.005[↩]
- Phadke R. Myopathology of adult and paediatric mitochondrial diseases. J Clin Med. 2017;6:64. doi: 10.3390/jcm6070064[↩]
- Quadir A, Pontifex CS, Lee Robertson H, Labos C, Pfeffer G. Systematic review and meta-analysis of cardiac involvement in mitochondrial myopathy. Neurol Genet. 2019;5:e339. doi: 10.1212/NXG.0000000000000339[↩]
- Yu Wai Man CY, Smith T, Chinnery PF, Turnbull DM, Griffiths PG. Assessment of visual function in chronic progressive external ophthalmoplegia. Eye (Lond). 2006 May;20(5):564-8. doi: 10.1038/sj.eye.6701924[↩][↩]
- Pitceathly RD, Morrow JM, Sinclair CD, Woodward C, Sweeney MG, Rahman S, Plant GT, Ali N, Bremner F, Davagnanam I, Yousry TA, Hanna MG, Thornton JS. Extra-ocular muscle MRI in genetically-defined mitochondrial disease. Eur Radiol. 2016 Jan;26(1):130-7. doi: 10.1007/s00330-015-3801-5[↩][↩]
- Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 2010;1797:113–28. doi: 10.1016/j.bbabio.2009.09.005[↩]
- McFarland R, Taylor RW, Turnbull DM. Mitochondrial disease–its impact, etiology, and pathology. Curr Top Develop Biol. 2007;77:113–55. doi: 10.1016/S0070-2153(06)77005-3[↩]
- Chinnery PF, Howell N, Andrews RM, Turnbull DM. Clinical mitochondrial genetics. J Med Genet. 1999 Jun;36(6):425-36. https://pmc.ncbi.nlm.nih.gov/articles/instance/1734386/pdf/v036p00425.pdf[↩]
- Greaves LC, Reeve AK, Taylor RW, Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–86. doi: 10.1002/path.3028[↩]
- Taylor RW, Schaefer AM, Barron MJ, McFarland R, Turnbull DM. The diagnosis of mitochondrial muscle disease. Neuromus Dis. 2004;14:237–45. doi: 10.1016/j.nmd.2003.12.004[↩]
- Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–43. doi: 10.1038/3804[↩]
- McFarland R, Clark KM, Morris AA, Taylor RW, Macphail S, Lightowlers RN, et al. Multiple neonatal deaths due to a homoplasmic mitochondrial DNA mutation. Nat Genet. 2002;30:145–6. doi: 10.1038/ng819[↩]
- Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–23. doi: 10.1002/ana.410430212[↩]
- Chinnery PF, Turnbull DM. Clinical features, investigation, and management of patients with defects of mitochondrial DNA. J Neurol Neurosug Psychiatry. 1997;63:559–63. doi: 10.1136/jnnp.63.5.559[↩]
- Kisler JE, Whittaker RG, McFarland R. Mitochondrial diseases in childhood: a clinical approach to investigation and management. Dev Med Child Neurol. 2010;52:422–33. doi: 10.1111/j.1469-8749.2009.03605.x[↩]
- Hill ME, Creed GA, McMullan TF, Tyers AG, Hilton-Jones D, Robinson DO, Hammans SR. Oculopharyngeal muscular dystrophy: phenotypic and genotypic studies in a UK population. Brain. 2001 Mar;124(Pt 3):522-6. doi: 10.1093/brain/124.3.522[↩][↩]
- Turner C, Hilton-Jones D. The myotonic dystrophies: diagnosis and management. J Neurol Neurosurg Psychiatry. 2010 Apr;81(4):358-67. doi: 10.1136/jnnp.2008.158261[↩]
- Machuca-Tzili L, Brook D, Hilton-Jones D. Clinical and molecular aspects of the myotonic dystrophies: a review. Muscle Nerve. 2005 Jul;32(1):1-18. doi: 10.1002/mus.20301[↩]
- Carlow TJ, Depper MH, Orrison WW Jr. MR of extraocular muscles in chronic progressive external ophthalmoplegia. AJNR Am J Neuroradiol. 1998 Jan;19(1):95-9. https://pmc.ncbi.nlm.nih.gov/articles/instance/8337335/pdf/9432164.pdf[↩]
- Barragán-Campos HM, Vallée JN, Lô D, Barrera-Ramírez CF, Argote-Greene M, Sánchez-Guerrero J, Estañol B, Guillevin R, Chiras J. Brain magnetic resonance imaging findings in patients with mitochondrial cytopathies. Arch Neurol. 2005 May;62(5):737-42. doi: 10.1001/archneur.62.5.737[↩]
- Vázquez-Justes D, Martín-Cucó A, Gallego-Sánchez Y, Vicente-Pascual M. WEBINO syndrome (wall-eyed bilateral internuclear ophthalmoplegia) secondary to ischemic stroke, about a case. Arch Soc Esp Oftalmol (Engl Ed). 2020 Apr;95(4):205-208. English, Spanish. doi: 10.1016/j.oftal.2019.12.012[↩]
- CAPOS Syndrome. https://eyewiki.org/CAPOS_Syndrome[↩]
- Le Cann M, Bouhour F, Viala K, Simon L, Tard C, Rossi C, Morel G, Lagrange E, Magy L, Créange A, Michaud M, Franques J, Echaniz-Laguna A, Antoine JC, Baron M, Arnulf B, Puma A, Delmont E, Maisonobe T, Leblond V, Roos-Weil D. CANOMAD: a neurological monoclonal gammopathy of clinical significance that benefits from B-cell-targeted therapies. Blood. 2020 Nov 19;136(21):2428-2436. doi: 10.1182/blood.2020007092[↩]
- Oculopharyngeal Muscular Dystrophy. https://eyewiki.org/Oculopharyngeal_Muscular_Dystrophy[↩]
- Brais B, Bouchard JP, Xie YG, Rochefort DL, Chrétien N, Tomé FM, Lafrenière RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codère F, Fardeau M, Rouleau GA. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998 Feb;18(2):164-7. doi: 10.1038/ng0298-164. Erratum in: Nat Genet 1998 Aug;19(4):404. Korcyn AD [corrected to Korczyn AD].[↩]
- Yu J, Deng J, Wang Z. Oculopharyngodistal myopathy. Curr Opin Neurol. 2022 Oct 1;35(5):637-644. doi: 10.1097/WCO.0000000000001089[↩]
- Ishiura H. Recent progress in oculopharyngodistal myopathy research from clinical and genetic viewpoints. J Neuromuscul Dis. 2025 Mar 3:22143602251319164. doi: 10.1177/22143602251319164[↩]
- Kirschner J, Bönnemann CG. The Congenital and Limb-Girdle Muscular Dystrophies: Sharpening the Focus, Blurring the Boundaries. Arch Neurol. 2004;61(2):189–199. doi:10.1001/archneur.61.2.189[↩]
- Evliyaoglu F, Karadag R, Burakgazi AZ. Ocular neuropathy in peripheral neuropathies. Muscle Nerve. 2012 Nov;46(5):681-6. https://www.interesjournals.org/articles/ocular-findings-in-muscular-dystrophies.pdf[↩]
- DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases. Muscle Nerve. 2006;34:265–83. doi: 10.1002/mus.20598[↩]
- Dimauro S, Rustin P. A critical approach to the therapy of mitochondrial respiratory chain and oxidative phosphorylation diseases. Biochim Biophys Acta. 2009;1792:1159–67. doi: 10.1016/j.bbadis.2008.10.015[↩]
- Karimi N, Kashkouli MB, Tahanian F, Abdolalizadeh P, Jafarpour S, Ghahvehchian H. Long-term results of palpebral fissure transfer with no lower eyelid spacer in chronic progressive external ophthalmoplegia. Am J Ophthalmol. 2022;234:99–107. doi: 10.1016/j.ajo.2021.07.027[↩]
- Ahn J., Kim N.J., Choung H.K., Hwang S.W., Sung M., Lee M.J., Khwarg S.I. Frontalis sling operation using silicone rod for the correction of ptosis in chronic progressive external ophthalmoplegia. Br. J. Ophthalmol. 2008;92:1685–1688. doi: 10.1136/bjo.2008.144816[↩]
- Rajabi MT, Tabatabaie SZ, Rajabi MB, Abrishami Y, Hosseini SS, Oestreicher J. Management of myogenic ptosis in chronic progressive external ophtalmoplegia. Iran J Neurol. 2014 Jul 4;13(3):185-7. https://pmc.ncbi.nlm.nih.gov/articles/PMC4240939[↩]
- Takahashi Y, Leibovitch I, Kakizaki H. Frontalis suspension surgery in upper eyelid blepharoptosis. Open Ophthalmol J. 2010 Dec 14;4:91-7. doi: 10.2174/1874364101004010091[↩][↩]
- Ahn J, Kim NJ, Choung HK, Hwang SW, Sung M, Lee MJ, Khwarg SI. Frontalis sling operation using silicone rod for the correction of ptosis in chronic progressive external ophthalmoplegia. Br J Ophthalmol. 2008 Dec;92(12):1685-8. doi: 10.1136/bjo.2008.144816[↩]
- Karimi N, Kashkouli MB, Tahanian F, Abdolalizadeh P, Jafarpour S, Ghahvehchian H. Long-term Results of Palpebral Fissure Transfer With No Lower Eyelid Spacer in Chronic Progressive External Ophthalmoplegia. Am J Ophthalmol. 2022 Feb;234:99-107. doi: 10.1016/j.ajo.2021.07.027[↩]
- Chronic Progressive External Ophthalmoplegia (CPEO). https://emedicine.medscape.com/article/1215103-overview#a2[↩]
- What is CPEO? https://umdf.org/cpeo[↩][↩]
- Barends M, Verschuren L, Morava E, Nesbitt V, Turnbull D, McFarland R. Causes of Death in Adults with Mitochondrial Disease. JIMD Rep. 2016;26:103-13. doi: 10.1007/8904_2015_449[↩][↩]




{kind=link}