Contents
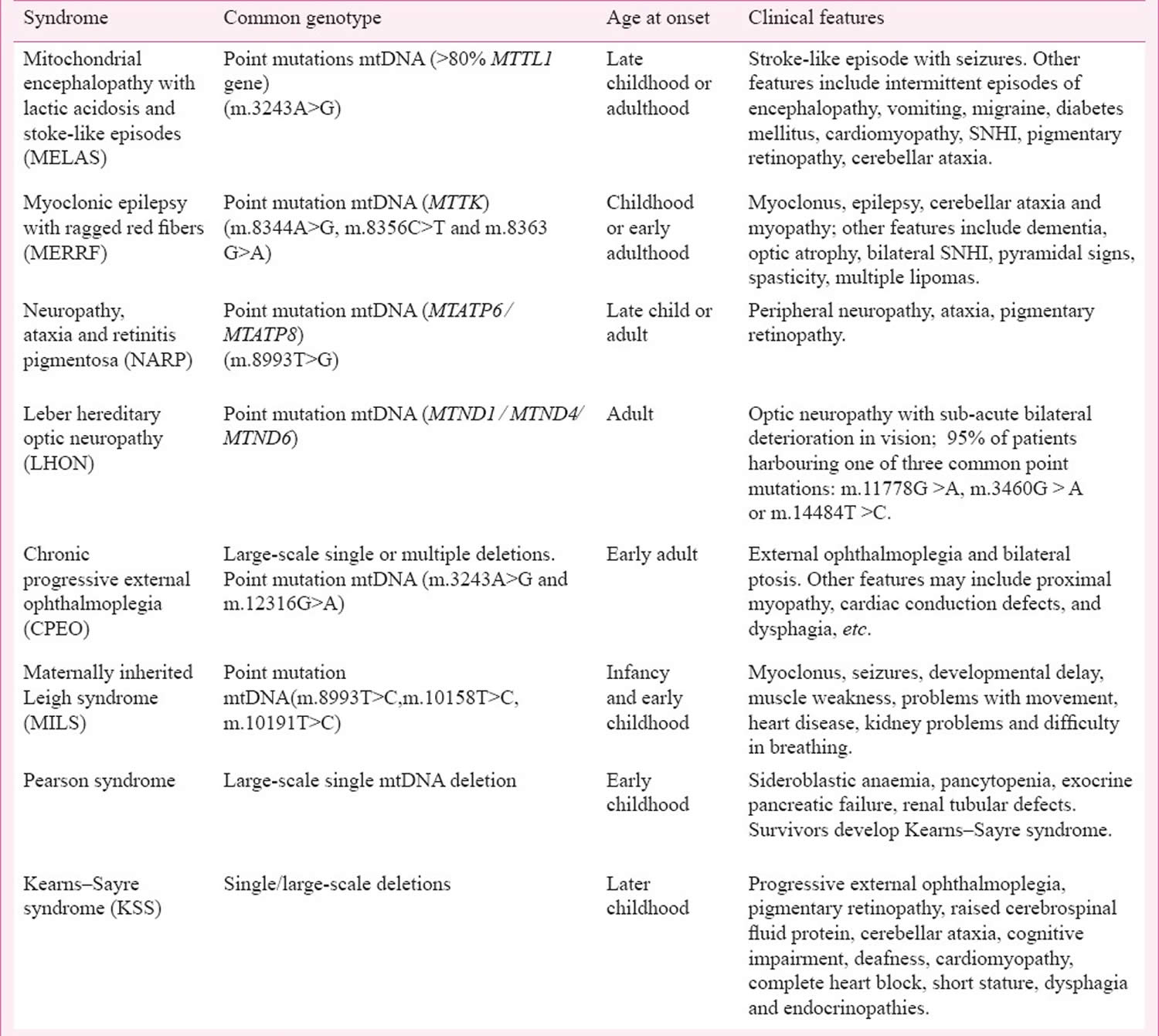
MELAS
MELAS also called MELAS syndrome or MELAS disease is an acronym for Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes is a rare progressive inherited mitochondrial disease that affects many of your body’s systems, particularly your brain and nervous system (encephalo-) and muscles (myopathy) 1, 2, 3, 4, 5, 6, 7, 8. MELAS also causes lactic acid to collect in your body (lactic acidosis) and may result in repeated events that are similar to strokes (stroke-like episodes) 1. MELAS signs and symptoms most often appear in childhood following a period of normal development, although they can begin at any age. Early symptoms may include muscle weakness and pain, trouble with physical activity, recurrent headaches, loss of appetite, vomiting, and seizures 1. Most affected individuals experience stroke-like episodes beginning before age 40 1. These stroke-like episodes often involve temporary muscle weakness on one side of the body (hemiparesis), altered consciousness, vision abnormalities, seizures, and severe headaches resembling migraines. Repeated stroke-like episodes can progressively damage the brain, leading to vision loss, problems with movement, memory loss and a loss of intellectual function (dementia) 1.
The exact incidence of MELAS is unknown. Mitochondrial diseases have an estimated incidence of 1 in 4,000 people 3. A study conducted on the adult Finnish population indicates a prevalence of 10.2 per 100,000 for the m.3243A>G mutation in the MT-TL1 gene 3. In Northern England, the prevalence of m.3243A>G mutation in the MT-TL1 gene in the adult population has been determined to be approximately 1 per 13,000 people. These figures might be underestimates. A comprehensive study of a large Caucasian population in 2006 revealed a prevalence of the MELAS people to be 236 per 100,000, significantly higher than previously reported 9.
Although both males and females are equally susceptible to MELAS disease, only women can pass MELAS on to their children as mitochondria are carried in the tails of sperm cells and are consequently shed outside the zygote during fertilization. No apparent racial predilection exists.
Most people with MELAS have a buildup of lactic acid in their bodies, a condition called lactic acidosis. Increased acidity in the blood can lead to vomiting, abdominal pain, extreme tiredness (fatigue), muscle weakness, and difficulty breathing. Less commonly, people with MELAS may experience involuntary muscle spasms (myoclonus), impaired muscle coordination (ataxia), hearing loss, heart and kidney problems, diabetes, nerve damage that affects sensation (peripheral neuropathy), short stature and hormonal imbalances 2, 4, 10, 1.
MELAS clinical presentations vary widely, MELAS signs and symptoms most often appear in childhood following a period of normal development, although they can begin at any age. MELAS signs and symptoms include stroke-like episodes, sensorineural hearing loss, and cognitive impairment associated with diffuse white matter injury. Less commonly, it can present with gastrointestinal signs and symptoms that include gastric perforation, ischemic colitis, segmental ileal paralysis, pseudo-obstruction, or megacolon. Endocrine dysfunction such as diabetes mellitus has also been reported in MELAS 11.
Eye signs and symptoms of MELAS include blindness over half the field of vision (hemianopia) and cortical blindness from stroke-like episodes, nystagmus, cataracts, CPEO, optic atrophy, salt and pepper pigmentary retinopathy, and macular degeneration 12.
The temporary stroke-like episodes of MELAS are characterized by nausea, vomiting, a migraine-like headache, encephalopathy, and focal seizures with or without neurological deficits. The exact pathogenic mechanism for stroke-like episodes is yet to be determined; however, three theories have been put forward.
- The first is insufficient energy due to mitochondrial dysfunction, supported by the increase in lactate peaks and decreased N-acetyl aspartate peaks of the occipital regions in brain magnetic resonance spectroscopy (MRS) 13.
- The second is nitric oxide (NO) deficiency, which usually regulates oxygenation and blood flow. This hypothesis is supported by a reduction in nitric oxide (NO) metabolites during acute attacks and an increase in nitric oxide (NO) synthase inhibitors in the COX-negative fibers of MELAS patients 14.
- The third theory is mitochondrial angiopathy, an accumulation of mitochondria in the smooth muscle cells and endothelial cells of small cerebral arteries leading to the narrowing of the lumen of blood vessels and reducing perfusion 15.
MELAS can result from mutations in one of several genes, including MT-TL1 (over 80% of MELAS cases), MT-ND5 (less than 10% of MELAS cases), MT-ND1, MT-TH, and MT-TV 16, 17, 1. These genes are found in the mitochondrial DNA (mtDNA). Some of the genes related to MELAS provide instructions for making proteins involved in normal mitochondrial function. These proteins are part of a large enzyme complex in mitochondria that helps convert oxygen, fats, and simple sugars to energy. Other genes associated with MELAS provide instructions for making molecules called transfer RNAs (tRNAs), which are chemical cousins of DNA. These molecules help assemble protein building blocks called amino acids into full-length, functioning proteins within mitochondria. Mutations in a particular transfer RNA gene, MT-TL1, cause more than 80 percent of all cases of MELAS 1. These mutations impair the ability of mitochondria to make proteins, use oxygen, and produce energy 18. Researchers have not determined how changes in mitochondrial DNA (mtDNA) lead to the specific signs and symptoms of MELAS. They continue to investigate the effects of mitochondrial gene mutations in different tissues, particularly in the brain.
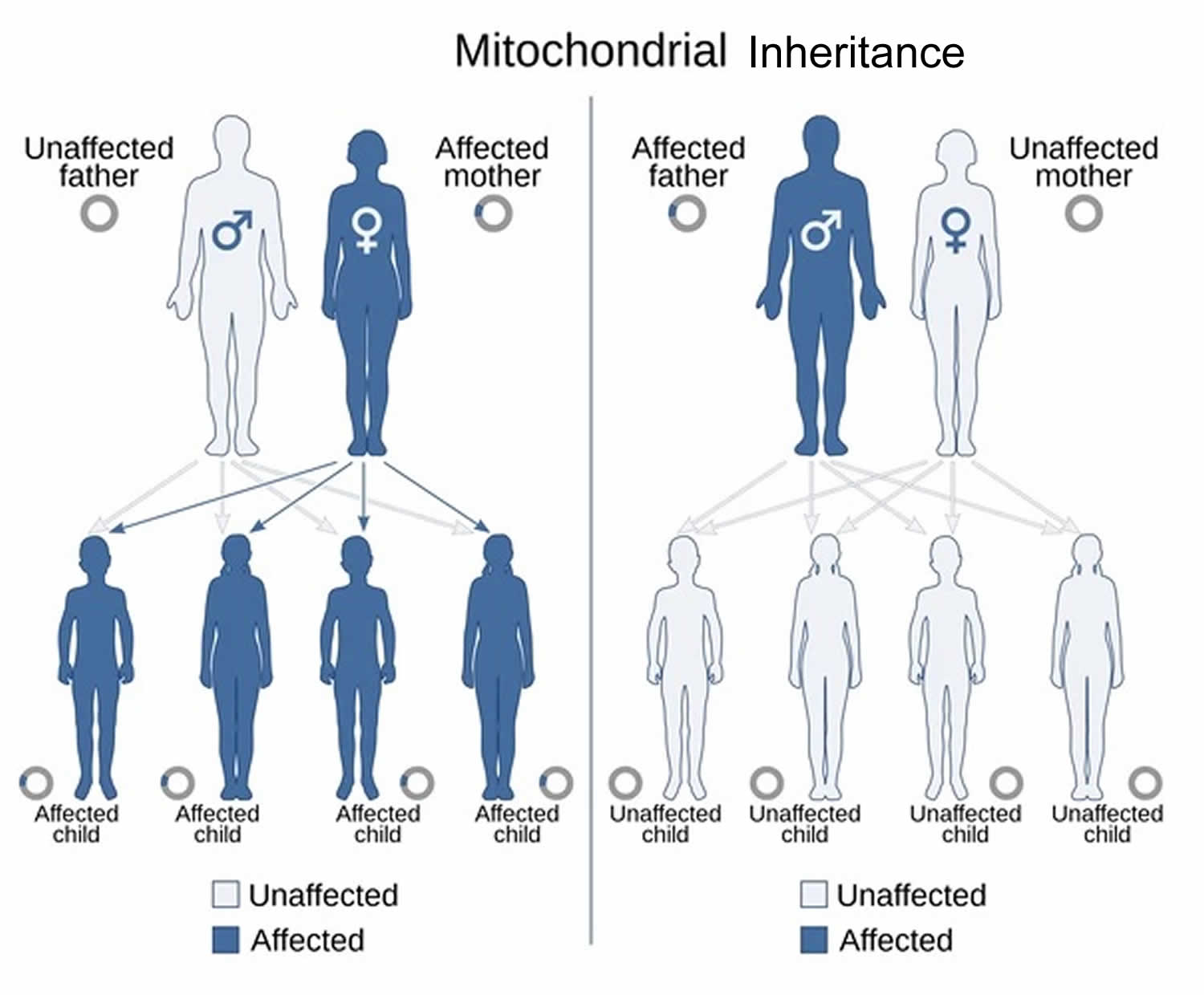
MELAS is inherited in a mitochondrial pattern, which is also known as maternal inheritance. Mitochondrial pattern pattern of inheritance (maternal inheritance) applies to genes contained in mitochondrial DNA (mtDNA). Because egg cells, but not sperm cells, contribute mitochondria to the developing embryo, children can only inherit disorders resulting from mtDNA mutations from their mother. These disorders can appear in every generation of a family and can affect both males and females, but fathers do not pass traits associated with changes in mtDNA to their children.
In most cases, people with MELAS inherit an altered mitochondrial gene from their mother. Less commonly, MELAS results from a new mutation in a mitochondrial gene and occurs in people with no family history of MELAS.
Tests to diagnose MELAS syndrome may include:
- Genetic tests
- Imaging tests, like a magnetic resonance imaging (MRI) scan
- Tests of cerebrospinal fluid, urine (pee) and blood
- Muscle biopsy (if needed)
Doctors use special brain scans (MRI) to check for stroke-like damage that doesn’t follow normal blood vessel patterns. Many patients also have high lactic acid levels in their blood (lactic acidosis) and show specific muscle tissue changes under a microscope known as “ragged red fibers” 2. Confirming MELAS diagnosis usually requires mitochondrial genetic testing 19, 20.
Currently there’s no cure or treatment that can slow or halt the progression of MELAS syndrome. MELAS treatment is directed to the specific symptoms that the affected person has and improving quality of life. It is important to have a team of specialists including neurologists, heart doctors (cardiologists), kidney doctors (nephrologists) and hormone specialists (endocrinologists) who should work together to develop a personalized treatment plan.
Your doctor may suggest 21, 3, 22, 23:
- Antiseizure medications: But you should avoid valproate. Many reports exist of valproate aggravating encephalopathy and seizures in patients with MELAS syndrome 3. The primary mechanism of valproate toxicity involves interference with mitochondrial beta-oxidation or direct mitochondrial toxicity, explaining the frequently observed elevated ammonia levels in patients taking valproate 24.
- Coenzyme Q10 and L-carnitine: These products may increase mitochondrial energy output and slow the progression of MELAS syndrome.
- L-arginine and L-citrulline: These products may decrease the number and effects of stroke-like episodes.
- Insulin or sulfonylureas: This is used to treat diabetes. Metformin should also be avoided because of its propensity to cause lactic acidosis 25.
- Your doctor may also suggest an exercise routine to strengthen your muscle and/or cochlear implants to help with hearing loss.
Figure 1. MELAS Syndrome
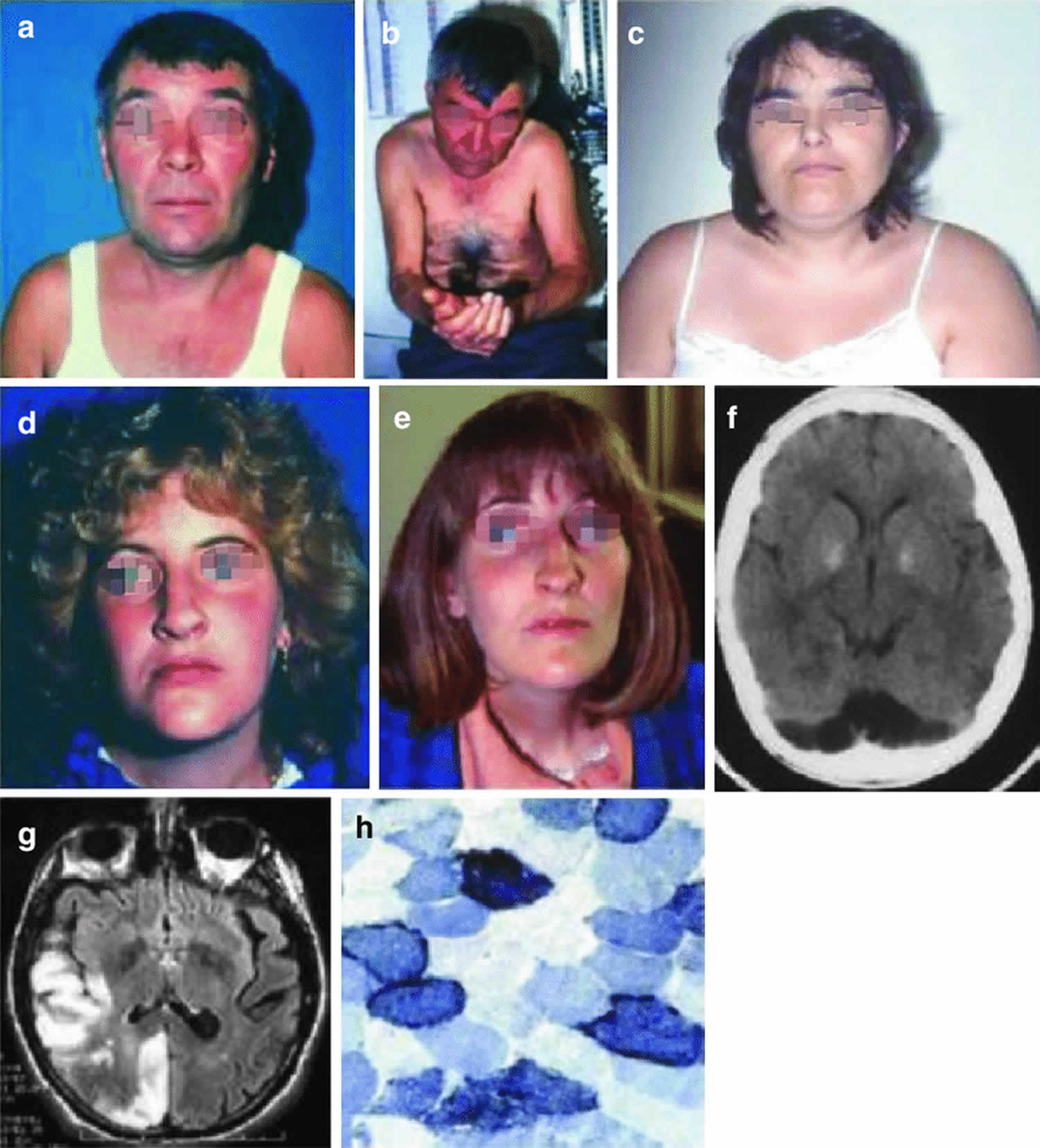
Footnotes: Three patients with MELAS syndrome. Patient 1 (a, b) had round face and palpebral ptosis, and after multiple strokes, he developed dementia and apraxia. His brain CT scan showed many lesions with nonvascular distribution (f). His niece (patient 4) (d, e) started with an ocular myopathy and died of dilated cardiomyopathy. Patient 3 (c) had round face, short stature, and limb weakness and developed an untreatable epileptic status. Muscle biopsy showed ragged-red fibers which strongly reacted with succinate dehydrogenase reaction (h). Brain MRI showed multiple stroke-like lesions (g) in occipital and temporoparietal areas.
[Source 26 ]Figure 2. MELAS Brain MRI
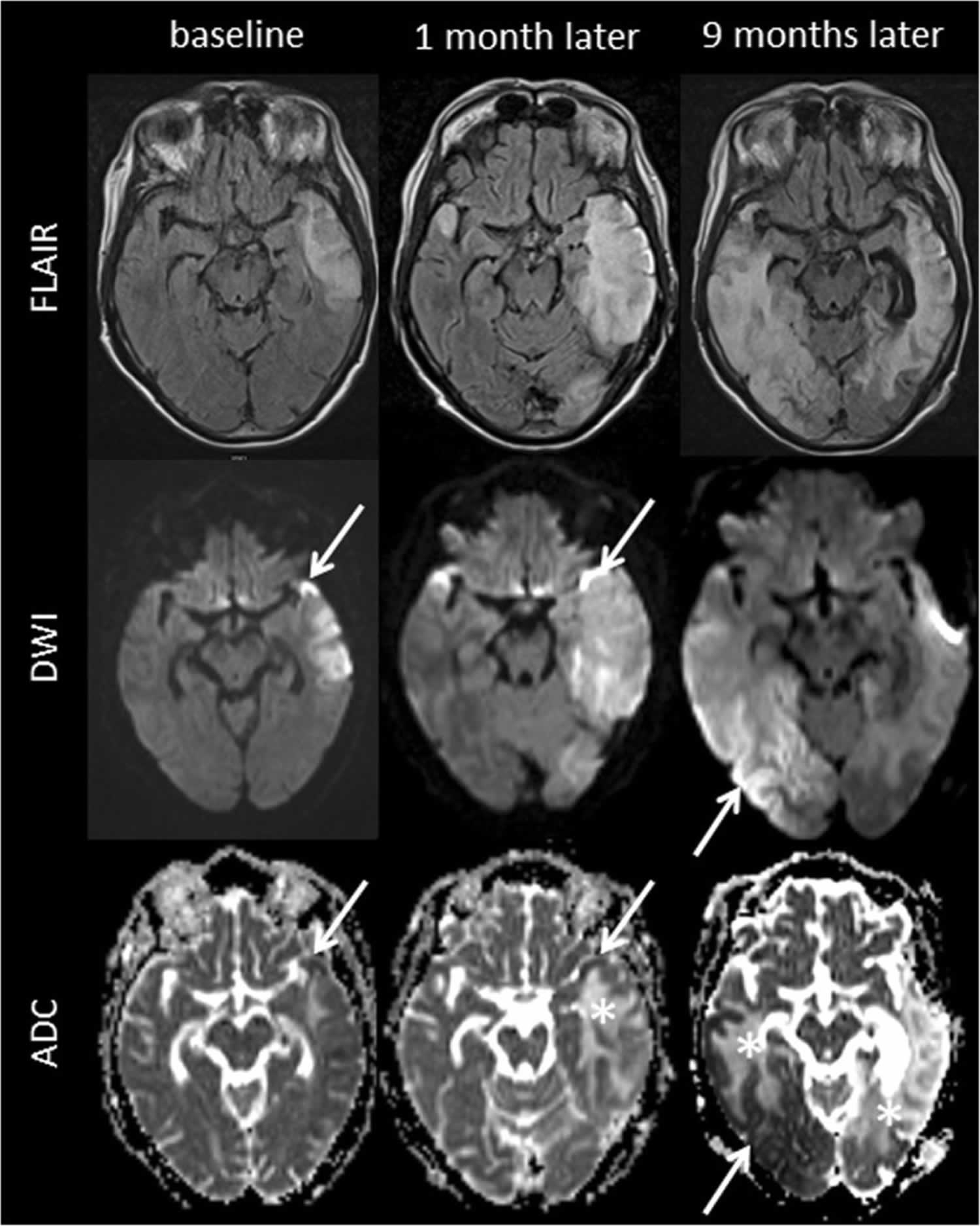
Footnotes: 63-year-old short stature female in whom the symptoms of MELAS were initially misdiagnosed as episodes of recurrent ischemic strokes with onset of acute aphasia, headache and a moderate right-sided hemiparesis. Medical history included type 2 diabetes, arterial hypertension, and past smoking. Whereas childhood and early adulthood were reportedly normal, she had developed hearing loss and diabetes at the age of 45 years. She was also diagnosed with cardiomyopathy that was initially thought to be of hypertensive aetiology, and had a history of chronic obstructive pulmonary disease (COPD) and renal insufficiency. Of note, the patient had two miscarriages, and one newborn child died within the first hours after birth. There was no history of previous frequent headaches.
MELAS MRI findings. Fluid attenuated inversion recovery (FLAIR), diffusion weighted imaging (DWI), and apparent diffusion coefficient (ADC) images at first presentation and until 9 months later are presented. FLAIR images demonstrate step-wise progressing FLAIR hyperintense oedema not corresponding to a vascular territory resulting in a local mass effect. DWI delineates both, i) cortical areas of restricted diffusion that are hyperintense on DWI and hypointense on ADC (arrows), as well as ii) subcortical vasogenic oedema indicated by ADC hyperintense areas (asterisks)
[Source 19 ]Figure 3. Muscle histochemistry
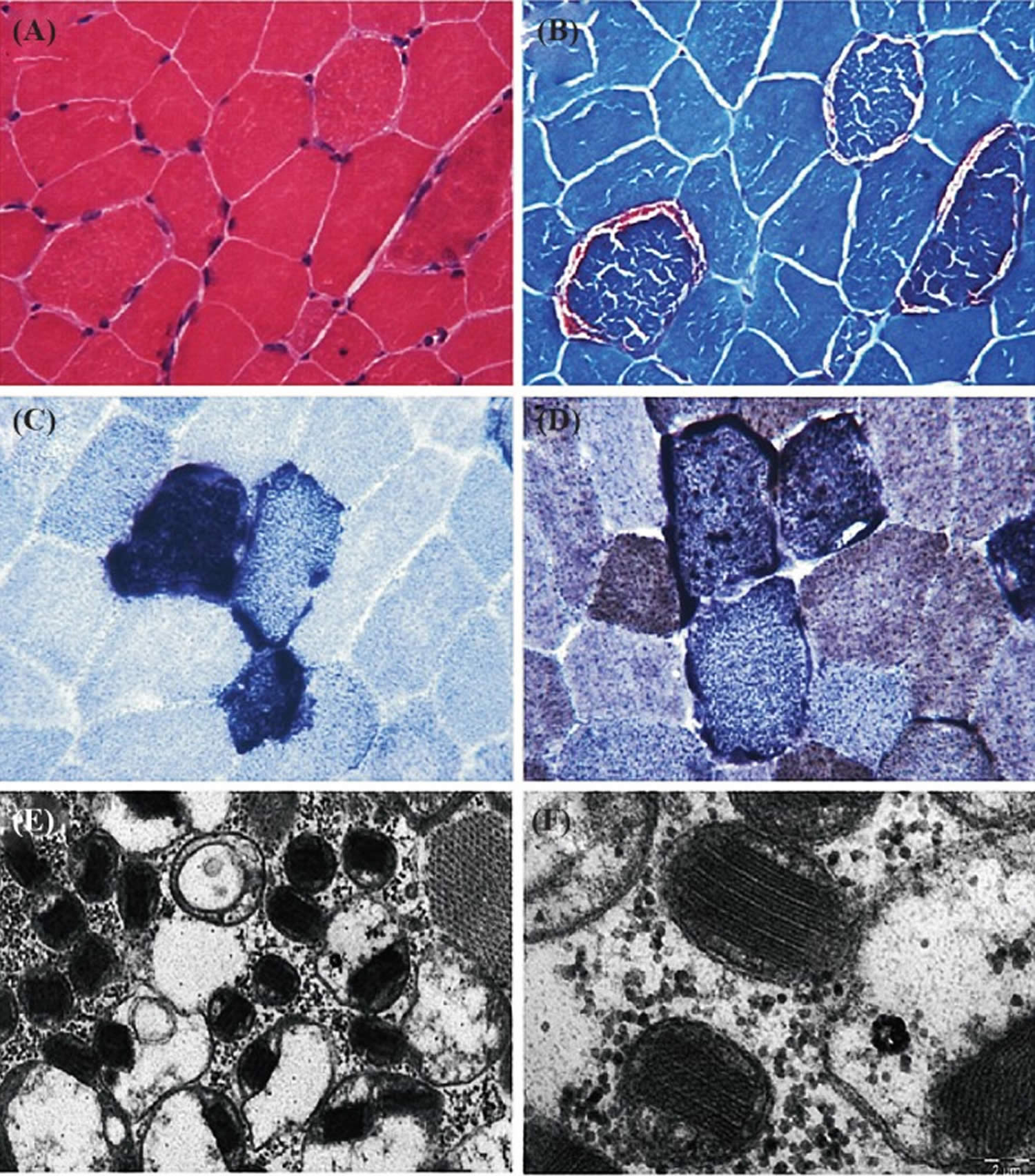
Footnotes: Tissue section showing the pathological abnormalities of mitochondrial disorders. (A) Hematoxylin and Eosin (HE) stain; (B) Red ragged fibers (RRF) appear on modified gomorie trichome stain suggesting abnormal subsarcolemmal accumulation of mitochondria; (C) Blue red fibers on succinate dehydrogenase (SDH) staining, suggesting an increased number of mitochondria; (D) COX staining shows absence of enzymes of respiratory chain. (E & F) Electron micrograph showing low and high magnification of mitochondria with paracrystalline inclusions (E x10000, F x40000).
[Source 27 ]Can MELAS syndrome be prevented?
No, you can’t prevent MELAS syndrome. But families with this inherited condition may want to consult a genetic counselor.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
What is mitochondria?
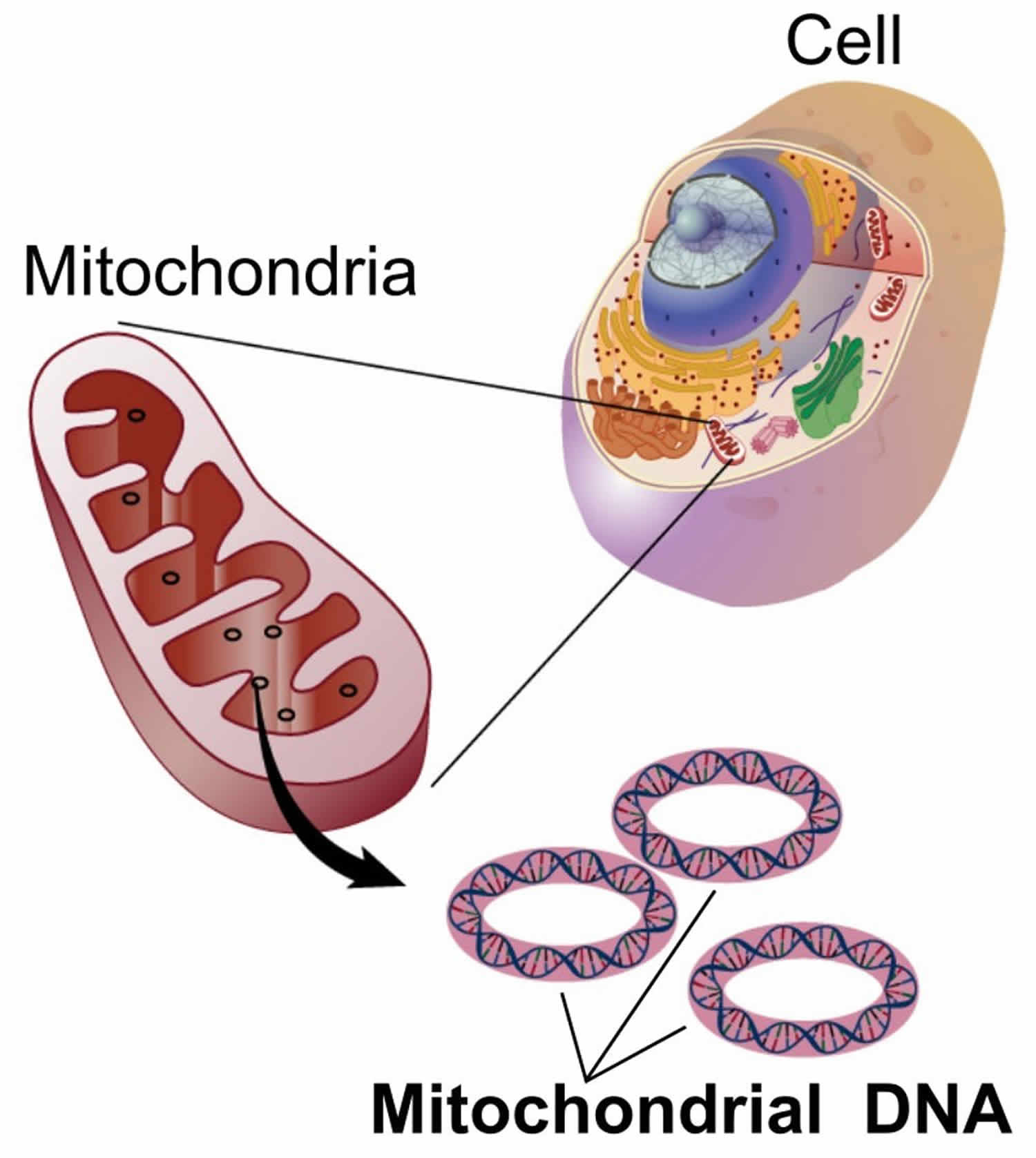
You have mitochondria present in every cell of your body except red blood cells. Mitochondria are membrane-bound cell organelles (mitochondrion, singular) within your cells, often called the “powerhouses” of the cell, in which a process called oxidative phosphorylation converts the energy from food you eat into a form called adenosine triphosphate (ATP) that your cells can use 28. Mitochondria use oxygen to break down glucose and other nutrients, releasing energy that is then stored in ATP (adenosine triphosphate). Your mitochondria also contain their own DNA known as mitochondrial DNA (mtDNA), which is essential for the normal function of these structures and is different from the DNA in your other cells nucleus.
The mitochondria in the cells throughout your body are responsible for creating more than 90% of the energy needed by your body to sustain life and support organ function. When mitochondria fail, less and less energy is generated within the cell. Cell injury and even cell death follow. If this process is repeated throughout the body, whole organ systems begin to fail – people get sick, and even die. The parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Chemical energy produced by the mitochondria is stored in a small molecule called adenosine triphosphate (ATP). Mitochondria contain their own small chromosomes. Generally, mitochondria, and therefore mitochondrial DNA, are inherited only from the mother. Problems with mitochondria, the structures that produce energy for all cells, have been linked to the development of Parkinson’s disease.
Mitochondria play a fundamental role in cell physiology; mitochondria organelles are involved in a variety of processes, including bioenergetics, various metabolic pathways, including crucial anabolic and catabolic reactions, such as ATP (adenosine triphosphate) synthesis, the tricarboxylic acid cycle (citric acid cycle or Kreb cycle), and biosynthetic processes, and govern fundamental cellular actions, including proliferation, immunity, and autophagy. Mitochondrial damage and malfunction have been related to the pathogenesis of a large number of human pathologies, such as mitochondrial diseases, neurodegenerative diseases, cancer, cardiovascular diseases, metabolic disorders, and aging. The participation of mitochondria in the redox equilibrium and redox signaling of the cell is also pivotal. Modification of the redox state and increased reactive oxygen species (ROS) production within mitochondria have major consequences for both mitochondrial and extramitochondrial processes and, ultimately, modulate fundamental cellular phenomena such as autophagy and apoptosis.
In people with mitochondrial disease, the parts of the body, such as the heart, brain, muscles and lungs, requiring the greatest amounts of energy are the most affected 29. Based upon recent epidemiological studies, mitochondrial disorders affect at least 1 in 8000 of the general population 30. Mitochondrial disease is difficult to diagnose, because it affects each individual differently. Symptoms can include seizures, strokes, severe developmental delays, inability to walk, talk, see, and digest food combined with a host of other complications. If three or more organ systems are involved, mitochondrial disease should be suspected.
Figure 4. Mitochondria cell
What is mitochondrial disease?
Mitochondrial diseases can affect almost any part of your body, including the cells of your:
- Brain.
- Nerves.
- Muscles.
- Kidneys.
- Heart.
- Liver.
- Eyes.
- Ears.
- Pancreas.
Figure 5. Mitochondrial diseases
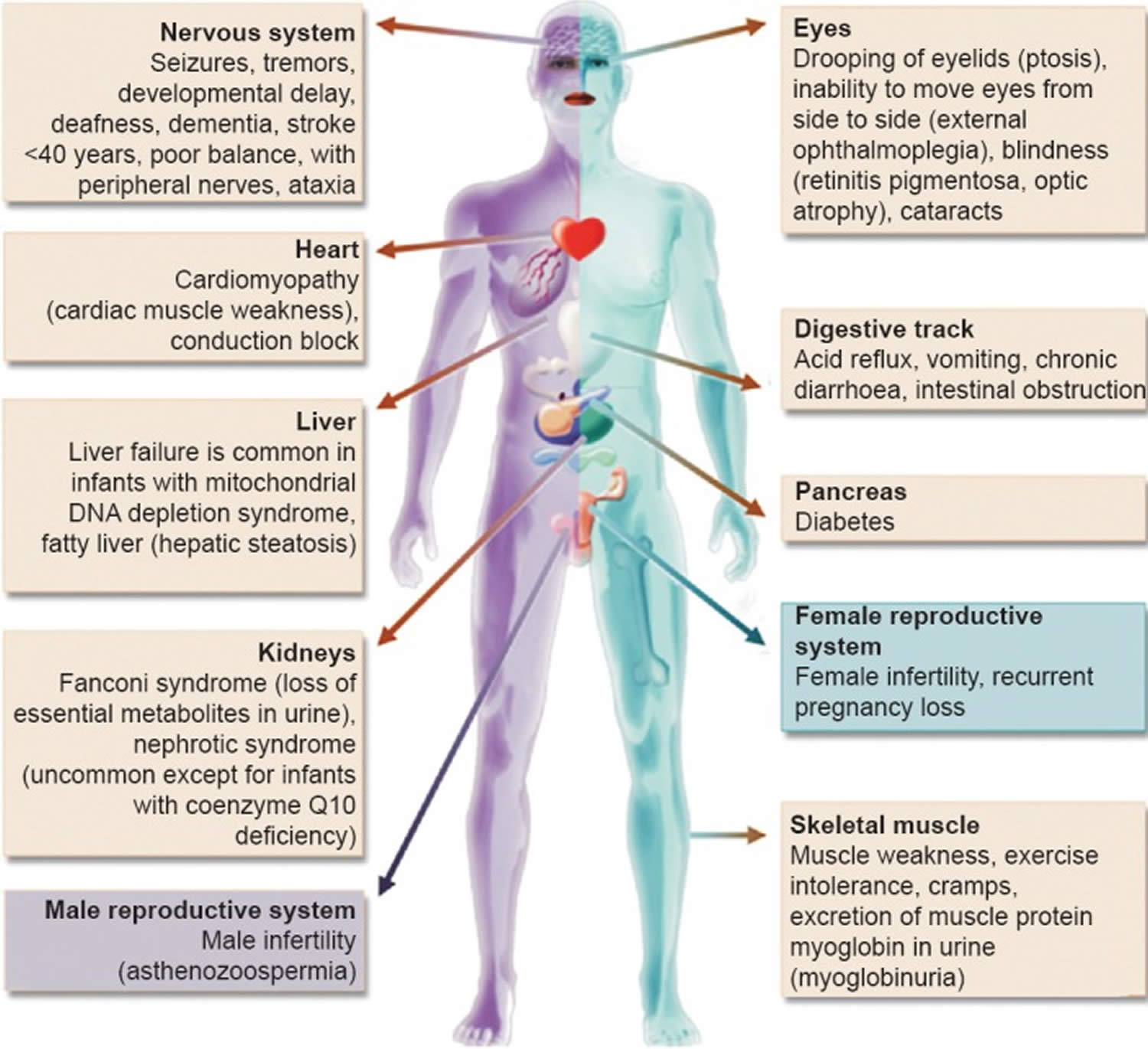
Footnote: Clinical features and the organs affected by mitochondrial diseases.
[Source 27 ]Figure 6. Mitochondrial diseases signs and symptoms
Are mitochondrial diseases difficult to diagnose?
Yes. Because mitochondrial diseases affect so many different organs and tissues of your body, and you may have many different symptoms, mitochondrial diseases can be difficult to diagnose. There’s no single laboratory test that can diagnose a mitochondrial disease. This is why a referral to a medical facility with physicians who specialize in these diseases is critical to making the diagnosis.
MELAS cause
MELAS is an inherited mitochondrial disorder caused by mutations (variants) that are thought to impair the mitochondrial DNA (mtDNA). MELAS mutations have been identified in the following mitochondrial genes 2, 3, 31, 32:
- MT-TL1 (>80% of MELAS cases) 17
- MT-ND5 (<10% of MELAS cases)
- MT-TC
- MT-TF
- MT-TH
- MT-TK
- MT-TL2
- MT-TQ
- MT-TV
- MT-TW
- MT-TS1
- MT-TS2
- MT-ND1
- MT-ND6
- MT-CO2
- MT-CO3
- MT-CYB
MELAS is primarily caused by mutations in mitochondrial DNA (mtDNA), with the most common being m.3243A>G in the MT-TL1 gene, which encodes transfer ribonucleic acid (tRNA) for leucine. A nucleotide substitution in transfer RNA (tRNA) is responsible for most cases of MELAS disease 3. One specific substitution, the m.3243A>G (A-to-G substitution at nucleotide 3243) in MT-TL1 gene is responsible for 80% of cases, whereas the m.3271T>C (T-to-C substitution at nucleotide 3271), accounts for the remaining cases 3.
Mitochondria are the cell structures that carry the body’s instructions for regulating energy production 33, 34. Genes for mitochondrial DNA (mtDNA) are inherited from the mother. This is because the mitochondrial DNA (mtDNA) in sperm cells is usually lost during fertilization. A mother with a gene mutation in mitochondrial DNA (mtDNA) will pass the mutation to all her children, and her daughters will pass the mutation to their children.
Normal mitochondrial genes and gene mutations can exist in the same cell, a situation known as heteroplasmy 22. The number of mitochondria with the gene mutation may be outnumbered by the number of mitochondria with a normal gene. Symptoms may not appear until a significant proportion of mitochondria have mtDNA with the gene mutation. The uneven distribution of normal genes and gene mutations in mitochondrial DNA (mtDNA) in different tissues can affect different organs in members of the same family. This can result in a variety of different symptoms in affected family members 3. The proportion of mutated mtDNA is directly correlated with the severity of clinical manifestations, as higher mutation loads typically result in more severe symptoms such as recurrent stroke-like episodes and progressive neurodegeneration 35, 36. This variability is also tissue-specific, with organs that have higher energy demands, such as the brain and muscles, being more severely affected when the mutation load surpasses a certain threshold. This phenomenon, known as the “threshold effect”, explains why MELAS can present with a wide range of symptoms across different patients, depending on the mutation load and the tissues affected 37, 2.
Another key aspect of MELAS genetics is its maternal inheritance pattern. Mitochondria are passed exclusively through the maternal line; therefore, all offspring of an affected mother will inherit the mtDNA mutation, but only daughters will transmit it to the next generation 5, 37, (El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [Updated 2018 Nov 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1233))
Additionally, MELAS is influenced by interactions between mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) which encodes most mitochondrial proteins, which can make the condition more complex and variable from person to person 2, 3. This interplay between mitochondrial DNA (mtDNA) and nuclear DNA (nDNA) is crucial because disruptions in this interaction can exacerbate mitochondrial dysfunction and further impair energy production in cells 38.
In addition to the genetic complexity, MELAS also demonstrates the evolutionary and functional implications of mtDNA mutations because variations in mitochondrial function can affect energy production and cellular homeostasis 5, 39, 36. These changes not only influence disease outcomes but may also impact long-term cellular adaptation, contributing to the broad spectrum of signs and symptoms seen in patients with MELAS. The combination of mtDNA mutations, heteroplasmy, and nuclear-mitochondrial interactions complicates the understanding and management of MELAS 37, 38.
Figure 7. MELAS maternal inheritance pattern
MELAS Pathophysiology
Mitochondrial dysfunction is the core driver of MELAS pathophysiology, predominantly caused by mitochondrial DNA (mtDNA) mutations, with the m.3243A>G mutation in the MT-TL1 gene being the most common 22. The m.3243A>G mutation in the MT-TL1 gene affects mitochondrial transfer RNA (tRNA) for leucine, thereby disrupting mitochondrial protein synthesis and impairing oxidative phosphorylation (OXPHOS) 22. Consequently, adenosine triphosphate (ATP) production is reduced, which particularly impacts energy-demanding tissues such as the brain, skeletal muscles, and heart 40, 41. The m.3243A>G mutation compromises the function of the electron transport chain (ETC), which forces cells to rely on anaerobic glycolysis, thereby leading to the accumulation of lactate and lactic acidosis, a hallmark of MELAS 42, 41. In addition to MT-TL1 gene, mutations in genes such as MT-ND that encode components of complex I in the electron transport chain (ETC) can also contribute to MELAS, illustrating the genetic heterogeneity of the syndrome 42, 43. A key factor influencing the severity of MELAS disease is heteroplasmy, in which mutated and normal mtDNA coexist within cells. The proportion of mutated mtDNA, known as the “mutation load”, determines the severity of clinical symptoms, with higher mutation loads leading to more severe manifestations 44, 42. This variation in mutation load across tissues, especially in high-energy-demanding organs, accounts for the multisystemic presentation of MELAS 40, 44. Mitochondrial dysfunction also leads to an overproduction of reactive oxygen species (ROS), contributing to oxidative stress and cellular damage 43, 44. Additionally, impaired calcium homeostasis, particularly in neurons, results in excitotoxicity and apoptosis, thereby contributing to neurodegeneration 43, 44. The accumulation of mutated mtDNA over generations due to the lack of recombination in mitochondrial inheritance further exacerbates the progression of MELAS, which highlights the chronic nature of mitochondrial dysfunction 42, 44. These complex interactions between genetic mutations, energy failure, oxidative stress, and calcium dysregulation drive the progressive and multisystemic nature of MELAS. These nuclear gene mutations disrupt essential processes such as mitochondrial deoxyribonucleic acid (mtDNA) replication, transcription, and translation, which can exacerbate mitochondrial dysfunction and add another layer of clinical variability to the disease. The interplay between nuclear and mitochondrial mutations highlights the complexity of MELAS, making its diagnosis and treatment more challenging 45.
MELAS signs and symptoms
MELAS (mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes) affects multiple organs and can appear at any age, though most people develop symptoms before age 20 2. The condition gets worse over time and the symptoms can vary widely among individuals 2. Children with MELAS usually experience normal early psychomotor development (development of an individual’s physical, cognitive, emotional, and social abilities) until symptoms emerge between the ages of 2 and 15 46. Although less frequent, infantile- and adult-onset are possible 46. The earliest signs in babies may include developmental delay, where a child may take longer than expected to reach milestones like sitting, walking, or talking and they may have trouble gaining weight and growing at a normal rate (failure to thrive) 47.
MELAS signs and symptoms may include 2, 46, 4:
- Neurological problems such as:
- Stroke-like episodes with weakness, paralysis, or numbness on one side of the body
- Focal neurological symptoms which are issues affecting specific parts of the body, resulting in weakness, vision problems, or trouble speaking
- These episodes do not follow normal stroke patterns
- Seizures that can be focal (affecting one part of the brain) or generalized (involving the whole brain)
- Headaches and migraines that are often severe and can trigger stroke-like episodes
- Cognitive decline (dementia) such as problems with memory, learning and thinking skills due to brain damage from repeated strokes
- Learning disabilities or delayed milestones in childhood
- Peripheral neuropathy with nerve problems leading to tingling, numbness, or pain in the hands and feet
- Encephalopathy, a condition that affects brain function, leading to confusion, difficulty thinking and reduced awareness
- Stroke-like episodes with weakness, paralysis, or numbness on one side of the body
- Psychiatric conditions such as:
- Depression
- Anxiety
- Bipolar disorder
- Psychosis
- Personality changes
- Attention problems such as attention deficit hyperactivity disorder (ADHD) which can make it harder for a child to focus, process information, or succeed in school
- Progressive loss of hearing, usually starting mildly
- Muscular problems (myopathy) such as:
- Muscle weakness, fatigue, trouble with physical activities and exercise intolerance
- Low muscle tone (hypotonia) with poor muscle strength, especially in young children
- Short stature when compared with family members
- Hormonal problems including:
- Diabetes that can be either type 1 diabetes or type 2 diabetes, appearing around age 38 on average
- Thyroid disorders in which some people may have an underactive (hypothyroidism) or overactive (hyperthyroidism) thyroid disease
- Growth hormone deficiency which can contribute to short stature
- Reproductive hormone problems that can lead to delayed puberty or other hormonal imbalances
- Heart problems including:
- Heart muscle disease (cardiomyopathy) which is the thickening or weakening of the heart muscle and can lead to heart failure
- Irregular heart rhythms (arrhythmias) in which conditions like Wolff-Parkinson-White syndrome may cause abnormal heartbeats
- High blood pressure in the lungs (pulmonary hypertension) that can make breathing difficult
- Lactic acidosis, a buildup of lactic acid in the blood, causing fatigue, nausea and breathing problems
- Stomach and intestinal problems including:
- Frequent vomiting and nausea, often triggered by stroke-like episodes or metabolic stress
- Intestinal problems that can include diarrhea, constipation and difficulty digesting food (intestinal pseudo-obstruction)
- Inflammation of the pancreas (pancreatitis), which can cause severe stomach pain
- Loss of appetite (anorexia) and a reduced desire to eat which can lead to weight loss or poor growth
- Frequent vomiting can happen without an obvious reason and may be triggered by metabolic stress
- Kidney problems (nephropathy) where some people may develop protein loss in the urine (proteinuria) or kidney disease
- Chronic anemia, a condition in which there are not enough red blood cells to carry oxygen
- Skin rashes such as vitiligo, small purple spots (purpura), excessive hair growth (hirsutism), or red skin patches (erythema)
The course of MELAS varies between individuals, but it generally worsens over time due to repeated stroke-like episodes and progressive neurological decline. Many affected people experience increasing disability, cognitive decline and muscle weakness as they age 2, 46, 4.
Table 1. MELAS Signs and Symptoms
| Frequency | Signs and Symptoms |
|---|---|
| ≥25% |
|
| 10%-24% |
|
| <10% |
|
Table 2. MELAS Additional Clinical Features
| Frequency | Clinical Features |
|---|---|
| ≥90% |
|
| 7%-89% |
|
| 50%-74% |
|
| 25%-49% |
|
| <25% |
|
Neurologic problems
- Stroke-like episodes. The stroke-like episodes associated with MELAS are a hallmark feature of the disease. Typically, patients exhibit an acute onset of neurological symptoms, such as hemiparesis (muscle weakness or partial paralysis on one side of the body that can affect the arms, legs and facial muscles), hemianopia (blindness over half the field of vision), or cortical blindness (a condition where vision is lost or impaired due to damage to the brain’s visual processing areas, specifically the occipital lobes). Initial stroke-like episodes may also involve vomiting and headaches that can last for several days. These episodes are considered “stroke-like” because there is no vascular occlusion, and the involvement does not correspond to any specific vascular territory 21. The apparent diffusion coefficient on MRI in individuals with MELAS may be increased or reveal a mixed pattern. Patients with an ischemic stroke show a decreased apparent diffusion coefficient on MRI. Acute MRI findings may also migrate and often resolve more quickly than changes of an ischemic stroke 48.
- Dementia affects intelligence, language, perception, attention, and memory function.
- Both the underlying neurologic dysfunction and the accumulating cortical injuries due to stroke-like episodes contribute to dementia.
- Executive function deficits have been observed despite the relative sparing of the frontal lobe on neuroimaging, indicating an additional diffuse neurodegenerative process in addition to the damage caused by the stroke-like episodes 49.
- Epilepsy
- Focal and primary generalized seizures can occur. Children with MELAS syndrome may experience seizures and visual abnormalities followed by hemiplegia. Seizure types may be tonic-clonic or myoclonic. Children with younger age of onset tend to have higher rates of drug-resistant epilepsy, leading to more severe clinical dysfunction 50.
- Primary generalized seizures in MELAS can occur in the context of normal neuroimaging or be accompanied by neuroimaging abnormalities including stroke-like episodes, white matter lesions, cortical atrophy, and corpus callosum agenesis or hypogenesis.
- Seizures can occur in MELAS as a manifestation of a stroke-like episode or independently, and may even induce a stroke-like episode 51.
- Migraine-Like Headaches in the form of recurrent attacks of severe pulsatile headaches with frequent vomiting are typical in individuals with MELAS and can precipitate stroke-like episodes. These headache episodes are often more severe during the stroke-like episodes 52.
- Hearing impairment due to sensorineural hearing loss is usually mild, insidiously progressive, and often an early clinical manifestation 51.
- Peripheral neuropathy is usually a chronic and progressive, sensorimotor, and distal polyneuropathy. Nerve conduction studies typically show an axonal or mixed axonal and demyelinating neuropathy 49.
- Early psychomotor development is usually normal, although developmental delay can occasionally occur.
- Psychiatric illnesses including depression, bipolar disorder, anxiety, psychosis, and personality changes can occur in MELAS 53.
- Additional findings associated with MELAS syndrome include vision loss due to optic atrophy, difficulties with night vision due to pigmentary retinopathy; excess thirst (polydipsia) and excessive urination (polyuria) as potential signs of diabetes; palpitations, and shortness of breath indicating heart conduction abnormalities such as Wolff-Parkinson-White syndrome or cardiomyopathy; numbness, tingling, and pain in the extremities accompanying peripheral neuropathy, a potential association with Alzheimer dementia linked to the m.3243 A>G variant.
Heart problems
- Both dilated and hypertrophic cardiomyopathy have been observed, however, the more typical is a non-obstructive concentric hypertrophy 49.
- Heart conduction abnormalities including Wolff-Parkinson-White syndrome has been reported occasionally 54.
Gastrointestinal problems
Recurrent or cyclic vomiting is common. Other manifestations include diarrhea, constipation, gastric dysmotility, intestinal pseudo-obstruction, recurrent pancreatitis, and failure to thrive 55.
Endocrine problems
- Diabetes mellitus occurs occasionally, with an average age of onset of 38 years. Diabetes can be type 1 or type 2. Individuals with type 2 diabetes can initially be treated with diet or sulfonylurea, although significant insulinopenia can develop and affected individuals may require insulin therapy 56.
- Short stature. Individuals with MELAS syndrome are typically shorter than their unaffected family members. Growth hormone deficiency has occasionally been reported 57 mutation. J Med Genet. 1996 Jul;33(7):621-2. https://pmc.ncbi.nlm.nih.gov/articles/instance/1050677/pdf/jmedgene00261-0093.pdf)).
- Hypothyroidism, hypogonadotropic hypogonadism, and hypoparathyroidism are infrequent manifestations 21.
Other signs and symptoms
- Kidney problems may include Fanconi proximal tubulopathy, proteinuria, and focal segmental glomerulosclerosis 58
- Pulmonary hypertension 59 gene (m.3243A>G). J Inherit Metab Dis. 2008 Dec;31 Suppl 3:497-503. doi: 10.1007/s10545-007-0735-3))
- Skin problems that may include vitiligo, small purple spots (purpura), excessive hair growth (hirsutism), or red skin patches (erythema) 60
- Chronic anemia 61
MELAS syndrome complications
Complications of MELAS syndrome may include 39:
- Failure to thrive and short stature
- Progressive intellectual deterioration possibly leading to dementia
- Development of psychiatric conditions such as depression with psychotic features, schizophrenia, or bipolar disorder
- Autism spectrum disorders
- Sensorineural hearing loss
- Endocrine dysfunction such as hypogonadism, diabetes, hypoparathyroidism, hypothyroidism, and hyperthyroidism
- Cardiomyopathy causing congestive heart failure
- Heart conduction defects causing sudden cardiac death
- Visual difficulties or cortical blindness (loss of vision)
- Focal segmental glomerulosclerosis leading to renal failure
- Acute kidney failure due to rhabdomyolysis
- Intestinal pseudoobstruction
- Pancreatitis
- Liver problems
- Aortic root dissection (although the relationship with the latter is currently unclear and requires further studies)
- Gait and balance issues
- Muscle issues, including spasms and lack of control.
MELAS diagnosis
MELAS diagnosis is made based on a combination of the characteristic clinical features, laboratory findings and genetic testing 3, 2, 22.
To diagnose MELAS your doctor may perform or recommend the following 2, 62:
- Genetic testing for MELAS gene mutation(s). The most common gene tested is MT-TL1. It is of note that the genetic test results alone cannot predict the diagnosis, because of variable signs and symptoms caused by the MELAS gene mutation(s), ranging from severe MELAS to diabetes, or deafness, or even asymptomatic.
- Diagnostic laboratory findings include demonstration of lactic acidosis. High lactic acid levels in the blood are often one of the first signs of MELAS disease, especially during a stroke-like episode. Lactic acid builds up when the mitochondria (the energy factories of cells) are not working properly, which happens in MELAS disease. Lactic acid levels are measured to help distinguish MELAS syndrome from other conditions that can cause strokes such as 3:
- High blood sugar (hyperglycemia)
- Low blood sugar (hypoglycemia)
- Metabolic disorders that affect how the body processes amino acids and fats
- People with MELAS typically have 3:
- High levels of lactic acid in the blood and cerebrospinal fluid (CSF)
- Increased lactic acid and pyruvate levels (another metabolic compound) after exercise
- A high lactate-to-pyruvate ratio, even though their oxygen levels are normal. In contrast, if lactic acid is high due to tissue damage like in a regular stroke, the lactate-to-pyruvate ratio is also high, but oxygen levels tend to be low. This difference helps to confirm a diagnosis of MELAS 3.
- Brain-imaging such as computed tomography (CT) scan or magnetic resonance imaging (MRI) to look for changes in the brain associated with MELAS disease. Since stroke-like episodes in MELAS can look like regular strokes so an MRI scan is essential to tell them apart. In MELAS, the affected brain areas do not match typical stroke locations, especially in children and teens. This helps confirm the diagnosis and rule out other causes of sudden neurological symptoms.
- Electroencephalogram (EEG or ‘brain wave’) testing for seizures and related brain issues.
- Muscle biopsy showing ragged red fibers (RRF), and respiratory chain analysis showing multiple partial defects. The diagnosis can be confirmed by molecular genetic testing.
- Heart evaluation with echocardiogram and electrocardiography (ECG) should be considered for all patients with MELAS to rule out heart changes and conduction defects 3.
Because MELAS is an inherited disorder, affected families should also receive genetic counseling.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
MELAS diagnostic criteria
Clinical diagnostic criteria for MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) have been published 7, 63. MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) should be suspected in individuals with the following clinical features.
Clinical Features
- Stroke-like episodes before the age of 40 years
- Acquired encephalopathy with seizures and/or dementia
- Recurrent headaches
- Muscle weakness and exercise intolerance
- Cortical vision loss
- Hemiparesis
- Recurrent vomiting
- Short stature
- Hearing impairment
- Normal early psychomotor development
- Peripheral neuropathy
- Learning disability.
1) A clinical diagnosis of MELAS can be made if the following 3 criteria are met 7:
- Stroke-like episodes before age 40 years
- Encephalopathy characterized by seizures and/or dementia
- Mitochondrial myopathy is evident by the presence of lactic acidosis and/or ragged-red fibers (RRFs) on muscle biopsy.
AND at least 2 of the following criteria are present:
- Normal early psychomotor development
- Recurrent headaches
- Recurrent vomiting episodes
2) A clinical diagnosis of MELAS can also be made in an individual with at least two category A AND two category B criteria 63:
- Category A criteria
- Headaches with vomiting
- Seizures
- Hemiplegia
- Cortical blindness
- Acute focal lesions on neuroimaging.
- Category B criteria
- High plasma or cerebrospinal fluid (CSF) lactate
- Mitochondrial abnormalities on muscle biopsy
- A MELAS-related pathogenic variant
Brain Imaging
Brain MRI
MRI findings of stroke-like episode exhibit stroke-like lesions that are usually differentiated from other pathologies by initially observing cortical and deep white matter lesions, in addition to occipital and parietal lobe lesions or lesions not confined to arterial territories. The most common MRI findings are cortical areas resembling multifocal infarcts in various stages of ischemic evolution, characterized by acute diffusion restriction, followed by subacute cortical laminar necrosis and eventual chronic volume loss 3. Notably, these findings do not conform to any known vascular territory. Initial lesions often manifest in the occipital or parietal lobes, eventually affecting the cerebellum, cerebral cortex, basal ganglia, and thalamus. Perfusion-weighted imaging (PWI) or arterial spin labeling (ASL) can also show hyperperfused lesions, and magnetic resonance spectroscopy exhibits lactate peaks 64. The well-described preferential distribution of lesions with restricted diffusion in the occipital cortical areas of individuals with MELAS syndrome is attributed to the high metabolic demand of these regions. The primary somatosensory cortex represents the second most common area of involvement associated with MELAS syndrome. The relatively symmetric cortical involvement observed on imaging in MELAS syndrome patients sets it apart from common stroke syndromes, reflecting symmetric areas of high metabolic demand 20.
Another distinctive finding in neuroimaging was reported in some cases of MELAS as cerebellar lesions stroke-like lesions 64, 65.
- During the stroke-like episodes, the affected areas:
- Slow spreading of the stroke-like lesions occurs in the weeks following the first symptoms, typically documented by T2-weighted MRI 66.
- Diffusion-weighted MRI shows increased apparent diffusion coefficient (ADC) in the stroke-like lesions of MELAS, in contrast to the decreased apparent diffusion coefficient (ADC) seen in ischemic strokes 67.
MR angiography (MRA) is usually normal and MR spectroscopy (MRS) shows decreased N-acetylaspartate signals and accumulation of lactate 49. Magnetic resonance spectroscopy (MRS) can detect metabolic abnormalities, including the lactate-to-creatine ratio in the muscle or brain and the decreased N-acetylaspartate-to-creatine ratio in stroke regions in the central nervous system. Magnetic resonance spectroscopy (MRS) may reveal regions of elevated lactate in the central nervous system, even when serum levels remain normal, and may identify an elevated lactate peak in both affected and unaffected brain areas 68.
Single-photon emission computed tomography: Single-photon emission computed tomography uses the tracer N-isopropyl-p-[123-I]-iodoamphetamine to identify strokes and monitor disease progression in individuals with MELAS.
Positron emission tomography (PET) may reveal a reduced cerebral metabolic rate for oxygen, an increased cerebral blood flow in cortical regions, and the preservation of the cerebral metabolic rate for glucose.
Head CT
- Computed tomography scans of the brain may reveal lucencies consistent with an infarction in individuals with MELAS. As time progresses, the common radiologic features of MELAS may include cerebral atrophy and basal ganglia calcification, a condition where calcium deposits build up in the basal ganglia, a part of the brain responsible for controlling movement, loss of brain tissue (atrophy) and multiple infarcts (areas of dead brain tissue caused by multiple strokes) 47.
Electromyography and Nerve Conduction Studies
Findings are consistent with a myopathic process, but neuropathy (nerve damage) may coexist. Neuropathy can be axonal or mixed axonal and demyelinating 69, 70.
Laboratory Findings
Lactic acidosis both in blood and cerebrospinal fluid (CSF). Lactic acidemia is very common. Cerebrospinal fluid (CSF) lactate is also elevated in most affected individuals. Lactic acidemia is not specific for MELAS syndrome as it can occur in other mitochondrial diseases, metabolic diseases, and systemic illness. Other situations unrelated to the diagnosis of MELAS syndrome in which lactate can be elevated are acute neurologic events such as seizure or stroke. On the other hand, lactate level can be normal in a minority of individuals with MELAS syndrome 71.
Elevated CSF protein rarely surpasses 100 mg/dL.
Muscle biopsy
Muscle biopsy is not required to make MELAS diagnosis; genetic testing is frequently used instead of muscle biopsy to establish the diagnosis.
- Ragged red fibers (RRFs) with the modified Gomori trichrome stain, which represent mitochondrial proliferation below the plasma membrane of the muscular fibers causing the contour of the muscle fiber to become irregular. These proliferated mitochondria also stain strongly with the succinate dehydrogenase (SDH) stain giving the appearance of ragged blue fibers.
Although ragged red fibers (RRFs) are present in many other mitochondrial diseases e.g., MERRF (myoclonic epilepsy with ragged red fibers), most of the ragged red fibers (RRFs) in MELAS stain positively with the cytochrome C oxidase (COX) histochemical stain, unlike other mitochondrial diseases where ragged red fibers do not react with cytochrome C oxidase (COX). - An overabundance of mitochondria in smooth muscle and endothelial cells of intramuscular blood vessels, best revealed with the succinate dehydrogenase (SDH) stain (“strongly succinate dehydrogenase-reactive blood vessels,” or SSVs)
- Respiratory chain studies on muscle tissue: typically multiple partial defects, especially involving complex I and/or complex IV. However, biochemical results can also be normal.
MELAS differential diagnosis
MELAS shares symptoms with many other conditions so other conditions must be ruled out before a diagnosis is made. Other mitochondrial disorders can have overlapping symptoms, so genetic testing is often needed to confirm MELAS. When a patient has a family history of mitochondrial disorders, doctors consider other possible conditions, including 3:
- Kearns-Sayre syndrome that causes vision problems, short stature, hearing loss and heart issues, making it different from MELAS
- MERRF (Myoclonic Epilepsy with Ragged-Red Fibers) is a rare, progressive mitochondrial disorder that primarily affects the muscles and nervous system. MERRF is similar to MELAS but includes sudden muscle jerks (myoclonic seizures) and may cause hearing and vision loss. MERRF is characterized by myoclonus (sudden muscle jerks), epilepsy, and ragged red fibers in muscle biopsies. MERRF is inherited through a maternal pattern, meaning the condition is passed down from a mother to her offspring. Genetic testing is needed to confirm the diagnosis.
- Leigh syndrome. Leigh syndrome is a inherited severe progressive neurodegenerative disorder that affects the central nervous system (brain and spinal cord), leading to a progressive degeneration of brain tissue that is characterized by progressive loss of mental and movement abilities (psychomotor regression) and resulting in early death within two to three years, often due to respiratory failure 72, 73. Leigh syndrome is the most common infantile form of mitochondrial disease, usually becomes apparent in infants and young children of 3 months to 2 years of age and affecting around 1 in 40,000 individuals 72, 73. A small number of individuals do not develop symptoms until adulthood or have symptoms that worsen more slowly. The first signs of Leigh syndrome seen in infancy are usually vomiting, diarrhea, and difficulty swallowing (dysphagia), which disrupts eating 73. These problems often result in an inability to grow and gain weight at the expected rate (failure to thrive). Severe muscle and movement problems are common in Leigh syndrome. Affected individuals may develop weak muscle tone (hypotonia), involuntary muscle contractions (dystonia), and problems with movement and balance (ataxia). Loss of sensation and weakness in the limbs (peripheral neuropathy), common in people with Leigh syndrome, may also make movement difficult 73. According to a meta-analysis by Chang et al. 74, the most common clinical features of Leigh syndrome were developmental delay, hypotonia, respiratory dysfunction, epilepsy, reduced feeding, and weakness.
Other conditions such as nutritional deficiencies, metabolic disorders and kidney disease may also cause similar symptoms, so genetic and metabolic tests are done to make an accurate diagnosis.
Other conditions that can cause strokes, since MELAS often causes stroke-like episodes, such as 2:
- Heart disease because conditions affecting the heart can lead to strokes due to blood clots
- Diseases of the arteries (carotid or vertebral diseases) because narrowed or damaged blood vessels can restrict blood flow to the brain
- Sickle cell disease is a group of inherited blood disorders where abnormal hemoglobin causes red blood cells to become rigid and sickle-shaped, leading to various health problems. The most common type is sickle cell anemia, where red blood cells are permanently sickle-shaped. This shape makes the cells sticky and inflexible, causing them to get stuck in blood vessels and block blood flow which increases the risk of strokes.
- Moyamoya disease is a rare progressive condition where the main arteries at the base of the brain narrow, leading to reduced blood flow and potential stroke or hemorrhage. The name “moyamoya” means “puff of smoke” in Japanese, referring to the appearance of tiny, compensatory blood vessels that develop around the narrowed arteries on a brain scan.
- Complicated migraines, as some types of migraines can mimic strokes and cause temporary neurological symptoms
- Fabry disease is a rare, inherited disorder caused by a deficiency of the alpha-galactosidase A enzyme, leading to the buildup of a fatty substance called globotriaosylceramide (Gb3) in the body. This buildup primarily affects the kidneys, heart, and nervous system, potentially causing serious complications like kidney failure, heart attack, and stroke.
MELAS treatment
Your doctor may suggest 21, 3, 22, 23:
- Anti-epileptic drugs: But you should avoid valproate. Many reports exist of valproate aggravating encephalopathy and seizures in patients with MELAS syndrome 3. The primary mechanism of valproate toxicity involves interference with mitochondrial beta-oxidation or direct mitochondrial toxicity, explaining the frequently observed elevated ammonia levels in patients taking valproate 24.
- Coenzyme Q10 and L-carnitine: These products may increase mitochondrial energy output and slow the progression of MELAS syndrome.
- L-arginine and L-citrulline: These products may decrease the number and effects of stroke-like episodes.
- Insulin or sulfonylureas: This is used to treat diabetes. Metformin should also be avoided because of its propensity to cause lactic acidosis 25.
- Your doctor may also suggest an exercise routine to strengthen your muscle and/or cochlear implants to help with hearing loss.
- Vaccines: People with MELAS syndrome should receive the necessary childhood vaccines, as well as the flu and pneumonia vaccines.
Since MELAS is associated with reduced levels of citrulline and arginine, which are nitric oxide (NO) precursors, and decreased nitric oxide (NO) that contributes to stroke-like episodes, supplement replacement with arginine was proposed. A systematic review by Argudo et al 75 concluded that the studies conducted showed promising results in managing stroke-like episodes. Acute phase management consists of giving an intravenous dose of 500 mg/kg/day or 10 g/m² in 24 hour for 3–5 days. Whereas chronically, 150–300 mg/kg/day (maximum of 500 mg) is used instead 76. A study conducted by Pek et al 16 using induced pluripotent stem cell-derived endothelial cells vouched for edaravone, a potent antioxidant, to be used for improving the vascular function in MELAS since it scavenges reactive oxygen species (ROS) and inhibits the inflammatory response in cerebrovascular diseases, which L-arginine and citrulline do not tackle. For treating epilepsy, levetiracetam is considered to be the first-line anticonvulsant in mitochondrial encephalomyopathy due to the mitochondrial toxicity of other anticonvulsant agents 77.
Table 3. Recommended Evaluations Following Diagnosis of MELAS
| System/Concern | Evaluation | Comment |
|---|---|---|
| Growth | Measurement of height & weight | |
| Eyes | Ophthalmology evaluation | To screen for ptosis, optic atrophy, pigmentary retinopathy, ophthalmoplegia, vision deficits |
| Ears | Audiology eval | To detect hearing loss |
| Cardiovascular | Echocardiogram | To screen for cardiomyopathy 1 |
| Electrocardiogram | To evaluate for conduction abnormalities 1 | |
| Renal | Urinalysis & urine amino acid analysis | To evaluate for renal tubulopathy |
| Musculoskeletal | Physical therapy or occupational therapy (OT) assessment | For individuals w/neurologic deficits |
| Neurologic | Neurologic eval | To assess for neurologic deficits 2 |
| Head MRI with magnetic resonance spectroscopy (MRS) | To evaluate for pathologic changes at baseline | |
| EEG | If seizures are suspected | |
| Neuropsychiatric testing | To assess cognitive abilities and for evidence of dementia | |
| Endocrinologic | Fasting serum glucose | To screen for diabetes mellitus 3 |
| Glucose tolerance test | ||
| Other | Consultation w/clinical geneticist &/or genetic counselor |
Footnotes:
1 Consider referral to a cardiologist.
2 Consider referral to a neurologist.
3 Consider referral to an endocrinologist.
MELAS syndrome medications and supplements
Treatments that may reduce symptoms and slow MELAS disease progression include 22:
- Medications for seizures such as anti-epileptic drugs. But you should avoid valproate. Many reports exist of valproate aggravating encephalopathy and seizures in patients with MELAS syndrome 3. The primary mechanism of valproate toxicity involves interference with mitochondrial beta-oxidation or direct mitochondrial toxicity, explaining the frequently observed elevated ammonia levels in patients taking valproate 24.
- Seizures in people with MELAS syndrome may be very difficult to treat.
- Status epilepticus (abnormally prolonged seizure or a series of seizures without a period of recovery between them) and elevated resting serum lactate levels are predictive of the development of drug-resistant epilepsy in MELAS 78. Poor seizure control is significantly associated with increased clinical disability. Early identification of high-risk patients for drug-resistant epilepsy could facilitate the development of more effective treatment plans 78.
- Supplements and vitamins that help with mitochondria function by providing nutrients that support energy production, including:
- Coenzyme Q10 (CoQ10): This is a natural compound that helps cells produce energy. It is often given in doses of 30 mg per kg of body weight per day.
- Riboflavin (Vitamin B2): Riboflavin (Vitamin B2) supports mitochondrial function, especially in people with specific enzyme deficiencies. Doses range from 50 to 400 mg per day.
- L-carnitine: This helps mitochondria process fats for energy. L-carnitine is given in doses of 50 to 100 mg per kg per day and it may improve energy levels and reduce toxic buildup in cells.
In addition to vitamins, other more advanced therapies may help:
- L-arginine and citrulline: These amino acids help widen blood vessels and may reduce the severity and frequency of stroke-like episodes.
- Taurine: This amino acid that may protect brain cells and improve mitochondrial function.
- Menadione (vitamin K3), phylloquinone (vitamin K1), and ascorbate (vitamin C), which are used to donate electrons to cytochrome C.
Diet therapy
- The ketogenic diet (high fat, low carbohydrate) may help some people with MELAS by providing an alternative energy source for the brain.
- Other specialized diets may be recommended depending on the patient’s specific metabolic needs.
Because these treatments can have side effects they must be carefully monitored by doctors.
In addition, treatment may include:
- Migraine treatments to help with severe headaches
- Hearing aids if hearing loss develops
- Physical therapy to help with muscle weakness and movement
- Speech and occupational therapy for people with learning difficulties or communication problems.
Table 4. MELAS syndrome treatment options
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Overall disease process | Coenzyme Q10
| May benefit some individuals 21 |
L-carnitine
| ||
Creatine
| ||
| Ptosis | Standard therapy | Eyelid “crutches,” blepharoplasty, or frontalis muscle-eyelid implantation could be considered. |
| Sensorineural hearing loss | Standard therapy | Cochlear implantation successful in some 79 |
| Cardiomyopathy | Standard pharmacologic therapy 80 | Per cardiologist |
| Cardiac conduction defects | ||
| Nephropathy | Standard therapy | |
| Exercise intolerance & weakness | Aerobic exercise 81 | |
| Stroke-like episodes | Arginine therapy 82, 83 | Physical & occupational therapy after acute phase, as appropriate |
| Seizures | Traditional anticonvulsant therapy | Avoid valproic acid. |
| Migraine headaches | Standard analgesics | |
| Diabetes mellitus (type 1 or type 2) | Dietary modification | May be particularly successful in thin individuals |
| Oral hypoglycemic agents | Avoid metformin. | |
| Insulin therapy 56 | Per endocrinologist |
Agents to Avoid
- Individuals with MELAS should avoid mitochondrial toxins such as: aminoglycoside antibiotics, linezolid, cigarettes, and alcohol. Valproic acid should be avoided in the treatment of seizures 84
- Metformin should also be avoided because of its propensity to cause lactic acidosis 49
- Dichloroacetate, which reduces lactate by activating the pyruvate dehydrogenase enzyme, should be avoided in MELAS syndrome. A study evaluating the effect of dichloroacetate in individuals with MELAS syndrome was terminated because of onset or worsening of peripheral neuropathy, indicating that dichloroacetate can be associated with peripheral nerve toxicity 85.
Pregnancy Management
Infertility may preclude pregnancy in some affected individuals. Women with MELAS syndrome should receive genetic counseling prior to pregnancy. During pregnancy, affected or at-risk women should be monitored for the development of diabetes mellitus and respiratory insufficiency, which may require therapeutic interventions 86.
MELAS prognosis
MELAS is relentlessly progressive disease progressing over years with episodic deterioration related to stroke-like events and the encephalomyopathy is usually severe and progresses to dementia 2, 3. The course varies from individual to individual 2. No available therapies slow or halt the progression of MELAS disease. The average survival is 17 years following the onset of seizures or neurological deficits 18.
In a cohort of 33 adults with the MT-TL1 gene mutation who were followed for three years, deterioration of sensorineural function, heart left-ventricular hypertrophy, EEG abnormalities, and overall severity were observed 87. In a natural history study of 31 individuals with MELAS and 54 symptomatic and asymptomatic obligate carrier relatives over a follow-up period of up to 10.6 years, neurologic examination, neuropsychological testing, and daily living scores significantly declined in all affected individuals with MELAS, whereas no significant deterioration occurred in carrier relatives 2. The death rate was more than 17-fold higher in fully symptomatic individuals compared to carrier relatives 2. The average observed age at death in the affected MELAS group was 34.5±19 years (range 10.2-81.8 years) 2. Of the deaths, 22% occurred in those younger than 18 years 2.
MELAS life expectancy
MELAS syndrome estimated overall median survival time based on fully symptomatic individuals was 16.9 years from onset of seizures or neurological deficits 18. A Japanese prospective cohort study of 96 individuals with MELAS disease confirmed a rapidly progressive course within a five-year interval, with 20.8% of affected individuals dying within a median time of 7.3 years from diagnosis 63.
- Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. https://medlineplus.gov/genetics/condition/mitochondrial-encephalomyopathy-lactic-acidosis-and-stroke-like-episodes[↩][↩][↩][↩][↩][↩][↩][↩]
- El-Hattab AW, Almannai M, Scaglia F. MELAS. 2001 Feb 27 [Updated 2018 Nov 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1233[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Pia S, Lui F. Melas Syndrome. [Updated 2024 Jan 25]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532959[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- MELAS. https://www.orpha.net/en/disease/detail/550[↩][↩][↩][↩]
- Tetsuka S., Ogawa T., Hashimoto R., Kato H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab. Brain Dis. 2021;36:2181–2193. doi: 10.1007/s11011-021-00772-x[↩][↩][↩]
- Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984 Oct;16(4):481-8. doi: 10.1002/ana.410160409[↩]
- Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, DiMauro S, Rowland LP. Melas: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992;2(2):125-35. doi: 10.1016/0960-8966(92)90045-8[↩][↩][↩][↩]
- Matsumoto J, Saver JL, Brennan KC, Ringman JM. Mitochondrial encephalomyopathy with lactic acidosis and stroke (MELAS). Rev Neurol Dis. 2005 Winter;2(1):30-4.[↩]
- Manwaring N, Jones MM, Wang JJ, Rochtchina E, Howard C, Mitchell P, Sue CM. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007 May;7(3):230-3. doi: 10.1016/j.mito.2006.12.004[↩]
- Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). https://omim.org/entry/540000[↩]
- Seed L.M., Dean A., Krishnakumar D., Phyu P., Horvath R., Harijan P.D. Molecular and neurological features of MELAS syndrome in paediatric patients: A case series and review of the literature. Mol. Genet. Genom. Med. 2022;10:e1955. doi: 10.1002/mgg3.1955[↩]
- Al-Enezi M., Al-Saleh H., Nasser M. Mitochondrial disorders with significant ophthalmic manifestations. Middle East Afr. J. Ophthalmol. 2008;15:81–86. doi: 10.4103/0974-9233.51998[↩]
- Weiduschat N., Kaufmann P., Mao X., Engelstad K.M., Hinton V., DiMauro S., De Vivo D., Shungu D. Cerebral metabolic abnormalities in A3243G mitochondrial DNA mutation carriers. Neurology. 2014;82:798–805. doi: 10.1212/WNL.0000000000000169[↩]
- El-Hattab A.W., Emrick L.T., Craigen W.J., Scaglia F. Citrulline and arginine utility in treating nitric oxide deficiency in mitochondrial disorders. Mol. Genet. Metab. 2012;107:247–252. doi: 10.1016/j.ymgme.2012.06.018[↩]
- Yoshida T., Ouchi A., Miura D., Shimoji K., Kinjo K., Sueyoshi T., Jonosono M., Rajput V. MELAS and reversible vasoconstriction of the major cerebral arteries. Intern. Med. 2013;52:1389–1392. doi: 10.2169/internalmedicine.52.0188[↩]
- Pek N.M.Q., Phua Q.H., Ho B.X., Pang J.K.S., Hor J.-H., An O., Yang H.H., Yu Y., Fan Y., Ng S.-Y., et al. Mitochondrial 3243A>G mutation confers pro-atherogenic and pro-inflammatory properties in MELAS iPS derived endothelial cells. Cell Death Dis. 2019;10:802. doi: 10.1038/s41419-019-2036-9[↩][↩]
- Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990 Dec 13;348(6302):651-3. doi: 10.1038/348651a0[↩][↩]
- Kaufmann P, Engelstad K, Wei Y, Kulikova R, Oskoui M, Sproule DM, Battista V, Koenigsberger DY, Pascual JM, Shanske S, Sano M, Mao X, Hirano M, Shungu DC, Dimauro S, De Vivo DC. Natural history of MELAS associated with mitochondrial DNA m.3243A>G genotype. Neurology. 2011 Nov 29;77(22):1965-71. doi: 10.1212/WNL.0b013e31823a0c7f[↩][↩][↩]
- Sinnecker T, Andelova M, Mayr M, Rüegg S, Sinnreich M, Hench J, Frank S, Schaller A, Stippich C, Wuerfel J, Bonati LH. Diagnosis of adult-onset MELAS syndrome in a 63-year-old patient with suspected recurrent strokes – a case report. BMC Neurol. 2019 May 8;19(1):91. doi: 10.1186/s12883-019-1306-6[↩][↩]
- Bhatia KD, Krishnan P, Kortman H, Klostranec J, Krings T. Acute Cortical Lesions in MELAS Syndrome: Anatomic Distribution, Symmetry, and Evolution. AJNR Am J Neuroradiol. 2020 Jan;41(1):167-173. doi: 10.3174/ajnr.A6325[↩][↩]
- El-Hattab AW, Adesina AM, Jones J, Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015 Sep-Oct;116(1-2):4-12. doi: 10.1016/j.ymgme.2015.06.004[↩][↩][↩][↩][↩]
- Na JH, Lee YM. Diagnosis and Management of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like Episodes Syndrome. Biomolecules. 2024 Nov 28;14(12):1524. doi: 10.3390/biom14121524[↩][↩][↩][↩][↩][↩][↩]
- Scaglia F., Northrop J.L. The mitochondrial myopathy encephalopathy, lactic acidosis with stroke-like episodes (MELAS) syndrome: A review of treatment options. CNS Drugs. 2006;20:443–464. doi: 10.2165/00023210-200620060-00002[↩][↩]
- Chaudhry N, Patidar Y, Puri V. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes unveiled by valproate. J Pediatr Neurosci. 2013 May;8(2):135-7. doi: 10.4103/1817-1745.117847[↩][↩][↩]
- Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008 Oct;1142:133-58. doi: 10.1196/annals.1444.011. Erratum in: Ann N Y Acad Sci. 2009 Apr;1161:601[↩][↩]
- Angelini, Corrado. (2018). Genetic Neuromuscular Disorders: A Case-Based Approach. doi:10.1007/978-3-319-56454-8.[↩]
- Khan NA, Govindaraj P, Meena AK, Thangaraj K. Mitochondrial disorders: challenges in diagnosis & treatment. Indian J Med Res. 2015 Jan;141(1):13-26. doi: 10.4103/0971-5916.154489[↩][↩][↩]
- Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A. 2011;108:13534–9. doi: 10.1073/pnas.1109263108[↩]
- Schapira AH. Mitochondrial disease. Lancet 2006;368(9529):70‐82.[↩]
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology 2008;63(1):35‐9.[↩]
- Ikeda T, Osaka H, Shimbo H, Tajika M, Yamazaki M, Ueda A, Murayama K, Yamagata T. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum Genome Var. 2018 Sep 4;5:25. doi: 10.1038/s41439-018-0026-6[↩]
- Niedermayr K, Pölzl G, Scholl-Bürgi S, Fauth C, Schweigmann U, Haberlandt E, Albrecht U, Zlamy M, Sperl W, Mayr JA, Karall D. Mitochondrial DNA mutation “m.3243A>G”-Heterogeneous clinical picture for cardiologists (“m.3243A>G”: A phenotypic chameleon). Congenit Heart Dis. 2018 Sep;13(5):671-677. doi: 10.1111/chd.12634[↩]
- Rahman J., Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560–2574. doi: 10.1016/S0140-6736(18)30727-X[↩]
- DiMauro S., Schon E.A. Mitochondrial respiratory-chain diseases. N. Engl. J. Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567[↩]
- El-Hattab A.W., Adesina A.M., Jones J., Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015;116:4–12. doi: 10.1016/j.ymgme.2015.06.004[↩]
- Li D., Liang C., Zhang T., Marley J.L., Zou W., Lian M., Ji D. Pathogenic mitochondrial DNA 3243A>G mutation: From genetics to phenotype. Front. Genet. 2022;13:951185. doi: 10.3389/fgene.2022.951185[↩][↩]
- Wallace D.C. Genetics: Mitochondrial DNA in evolution and disease. Nature. 2016;535:498–500. doi: 10.1038/nature18902[↩][↩][↩]
- Nitsch L., Lareau C.A., Ludwig L.S. Mitochondrial genetics through the lens of single-cell multi-omics. Nat. Genet. 2024;56:1355–1365. doi: 10.1038/s41588-024-01794-8[↩][↩]
- Fan H.C., Lee H.F., Yue C.T., Chi C.S. Clinical Characteristics of Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Life. 2021;11:1111. doi: 10.3390/life11111111[↩][↩]
- Pickett S.J., Grady J.P., Ng Y.S., Gorman G.S., Schaefer A.M., Wilson I.J., Cordell H.J., Turnbull D.M., Taylor R.W., McFarland R. Phenotypic heterogeneity in m.3243A>G mitochondrial disease: The role of nuclear factors. Ann. Clin. Transl. Neurol. 2018;5:333–345. doi: 10.1002/acn3.532[↩][↩]
- Barros C.D.S., Coutinho A., Tengan C.H. Arginine Supplementation in MELAS Syndrome: What Do We Know about the Mechanisms? Int. J. Mol. Sci. 2024;25:3629. doi: 10.3390/ijms25073629[↩][↩]
- Zheng H., Zhang X., Tian L., Liu B., He X., Wang L., Ding S., Guo Y., Cai J. Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes with an MT-TL1 m.3243A>G point mutation: Neuroradiological features and their implications for underlying pathogenesis. Front. Neurosci. 2022;16:1028762. doi: 10.3389/fnins.2022.1028762[↩][↩][↩][↩]
- Scholle L.M., Zierz S., Mawrin C., Wickenhauser C., Urban D.L. Heteroplasmy and Copy Number in the Common m.3243A>G Mutation-A Post-Mortem Genotype-Phenotype Analysis. Genes. 2020;11:212. doi: 10.3390/genes11020212[↩][↩][↩]
- Ikeda T., Osaka H., Shimbo H., Tajika M., Yamazaki M., Ueda A., Murayama K., Yamagata T. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum. Genome Var. 2018;5:25. doi: 10.1038/s41439-018-0026-6[↩][↩][↩][↩][↩]
- Chakrabarty S., Govindaraj P., Sankaran B.P., Nagappa M., Kabekkodu S.P., Jayaram P., Mallya S., Deepha S., Ponmalar J.N.J., Arivinda H.R., et al. Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. J. Neurol. 2021;268:2192–2207. doi: 10.1007/s00415-020-10390-9[↩]
- MELAS Syndrome. https://rarediseases.org/rare-diseases/melas-syndrome[↩][↩][↩][↩]
- Watchalotone S, McDonald H, Degolier J, Schlangen A, Ahmed I. Radiologic Findings in a Patient With Mitochondrial Encephalomyopathy With Lactic Acidosis and Stroke-Like Episodes Syndrome: A Case Report. Cureus. 2024 Dec 7;16(12):e75268. doi: 10.7759/cureus.75268[↩][↩]
- Alves CAPF, Zandifar A, Peterson JT, Tara SZ, Ganetzky R, Viaene AN, Andronikou S, Falk MJ, Vossough A, Goldstein AC. MELAS: Phenotype Classification into Classic-versus-Atypical Presentations. AJNR Am J Neuroradiol. 2023 May;44(5):602-610. doi: 10.3174/ajnr.A7837[↩]
- Sproule DM, Kaufmann P. Mitochondrial encephalopathy, lactic acidosis, and strokelike episodes: basic concepts, clinical phenotype, and therapeutic management of MELAS syndrome. Ann N Y Acad Sci. 2008 Oct;1142:133-58. doi: 10.1196/annals.1444.011. Erratum in: Ann N Y Acad Sci. 2009 Apr;1161:601.[↩][↩][↩][↩][↩][↩]
- Li J, Zhang W, Cui Z, Li Z, Jiang T, Meng H. Epilepsy Associated With Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front Neurol. 2021 Jun 11;12:675816. doi: 10.3389/fneur.2021.675816[↩]
- Finsterer J, Zarrouk-Mahjoub S. Focal and Generalized Seizures May Occur in Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes (MELAS) Patients. J Child Neurol. 2015 Oct;30(11):1553-4. doi: 10.1177/0883073814567539[↩][↩]
- Ohno K, Isotani E, Hirakawa K. MELAS presenting as migraine complicated by stroke: case report. Neuroradiology. 1997 Nov;39(11):781-4. doi: 10.1007/s002340050505[↩]
- Anglin RE, Garside SL, Tarnopolsky MA, Mazurek MF, Rosebush PI. The psychiatric manifestations of mitochondrial disorders: a case and review of the literature. J Clin Psychiatry. 2012 Apr;73(4):506-12. doi: 10.4088/JCP.11r07237[↩]
- Sproule DM, Kaufmann P, Engelstad K, Starc TJ, Hordof AJ, De Vivo DC. Wolff-Parkinson-White syndrome in Patients With MELAS. Arch Neurol. 2007 Nov;64(11):1625-7. doi: 10.1001/archneur.64.11.1625[↩]
- Fujii A, Yoneda M, Ohtani M, Nakagawa H, Kumano T, Hayashi K, Muramatsu A, Takabatake S, Ibi T, Sahashi K, Azuma T, Kuriyama M. Gastric dysmotility associated with accumulation of mitochondrial A3243G mutation in the stomach. Intern Med. 2004 Dec;43(12):1126-30. doi: 10.2169/internalmedicine.43.1126[↩]
- Maassen JA, ‘T Hart LM, Van Essen E, Heine RJ, Nijpels G, Jahangir Tafrechi RS, Raap AK, Janssen GM, Lemkes HH. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004 Feb;53 Suppl 1:S103-9. doi: 10.2337/diabetes.53.2007.s103[↩][↩]
- Yorifuji T, Kawai M, Momoi T, Sasaki H, Furusho K, Muroi J, Shimizu K, Takahashi Y, Matsumura M, Nambu M, Okuno T. Nephropathy and growth hormone deficiency in a patient with mitochondrial tRNA(Leu(UUR[↩]
- Hotta O, Inoue CN, Miyabayashi S, Furuta T, Takeuchi A, Taguma Y. Clinical and pathologic features of focal segmental glomerulosclerosis with mitochondrial tRNALeu(UUR) gene mutation. Kidney Int. 2001 Apr;59(4):1236-43. doi: 10.1046/j.1523-1755.2001.0590041236.x[↩]
- Sproule DM, Dyme J, Coku J, de Vinck D, Rosenzweig E, Chung WK, De Vivo DC. Pulmonary artery hypertension in a child with MELAS due to a point mutation of the mitochondrial tRNA((Leu[↩]
- Kubota Y, Ishii T, Sugihara H, Goto Y, Mizoguchi M. Skin manifestations of a patient with mitochondrial encephalomyopathy with lactic acidosis and strokelike episodes (MELAS syndrome). J Am Acad Dermatol. 1999 Sep;41(3 Pt 1):469-73. doi: 10.1016/s0190-9622(99)70123-4[↩]
- Finsterer J. Chronic anemia as a manifestation of MELAS syndrome. Rev Invest Clin. 2011 Jan-Feb;63(1):100-3.[↩]
- Cohen BH, Chinnery PF, Copeland WC. POLG-Related Disorders. 2010 Mar 16 [Updated 2024 Feb 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK26471[↩]
- Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, Kakuma T, Koga Y; Taro Matsuoka for MELAS Study Group in Japan. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012 May;1820(5):619-24. doi: 10.1016/j.bbagen.2011.03.015[↩][↩][↩]
- Cheng W., Zhang Y., He L. MRI Features of Stroke-Like Episodes in Mitochondrial Encephalomyopathy with Lactic Acidosis and Stroke-Like Episodes. Front. Neurol. 2022;13:843386. doi: 10.3389/fneur.2022.843386[↩][↩]
- Ng Y.S., Bindoff L.A., Gorman G.S., Horvath R., Klopstock T., Mancuso M., Martikainen M.H., Mcfarland R., Nesbitt V., Pitceathly R.D.S., et al. Consensus-based statements for the management of mitochondrial stroke-like episodes. Wellcome Open Res. 2019;4:201. doi: 10.12688/wellcomeopenres.15599.1[↩]
- Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread of the stroke-like lesions in MELAS. Neurology. 2003 Nov 11;61(9):1238-44. doi: 10.1212/01.wnl.0000091888.26232.fe[↩]
- Kolb SJ, Costello F, Lee AG, White M, Wong S, Schwartz ED, Messé SR, Ellenbogen J, Kasner SE, Galetta SL. Distinguishing ischemic stroke from the stroke-like lesions of MELAS using apparent diffusion coefficient mapping. J Neurol Sci. 2003 Dec 15;216(1):11-5. doi: 10.1016/s0022-510x(03)00218-1[↩]
- Möller HE, Kurlemann G, Pützler M, Wiedermann D, Hilbich T, Fiedler B. Magnetic resonance spectroscopy in patients with MELAS. J Neurol Sci. 2005 Mar 15;229-230:131-9. doi: 10.1016/j.jns.2004.11.014[↩]
- Kärppä M, Syrjälä P, Tolonen U, Majamaa K. Peripheral neuropathy in patients with the 3243A>G mutation in mitochondrial DNA. J Neurol. 2003 Feb;250(2):216-21. doi: 10.1007/s00415-003-0981-8[↩]
- Kaufmann P, Pascual JM, Anziska Y, Gooch CL, Engelstad K, Jhung S, DiMauro S, De Vivo DC. Nerve conduction abnormalities in patients with MELAS and the A3243G mutation. Arch Neurol. 2006 May;63(5):746-8. doi: 10.1001/archneur.63.5.746[↩]
- Hirano M, Pavlakis SG. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS): current concepts. J Child Neurol. 1994 Jan;9(1):4-13. doi: 10.1177/088307389400900102[↩]
- Saini A.G., Chatterjee D., Bhagwat C., Vyas S., Attri S.V. Leigh syndrome in an infant: Autopsy and histopathology findings. Autops. Case Rep. 2021;11:e2021334. doi: 10.4322/acr.2021.334[↩][↩]
- Leigh syndrome. https://medlineplus.gov/genetics/condition/leigh-syndrome[↩][↩][↩][↩]
- Chang X., Wu Y., Zhou J., Meng H., Zhang W., Guo J. A meta-analysis and systematic review of Leigh syndrome: Clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine. 2020;99:e18634. doi: 10.1097/MD.0000000000018634[↩]
- Argudo J.M., Moncayo O.M.A., Insuasti W., Garofalo G., Aguirre A.S., Encalada S., Villamarin J., Oña S., Tenemaza M.G., Eissa-Garcés A., et al. Arginine for the Treatment of Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-Like Episodes: A Systematic Review. Cureus. 2022;14:e32709. doi: 10.7759/cureus.32709[↩]
- Pérez-Cruz E., González-Rivera C., Valencia-Olvera L.d.C.G. Immunonutrition for the acute treatment of MELAS syndrome. Endocrinología. Diabetes Nutr. 2022;69:144–148. doi: 10.1016/j.endien.2022.02.006[↩]
- Li J., Zhang W., Cui Z., Li Z., Jiang T., Meng H. Epilepsy Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Front. Neurol. 2021;12:675816. doi: 10.3389/fneur.2021.675816[↩]
- Gao R, Gu L, Zuo W, Wang P. Comprehensive predictors of drug-resistant epilepsy in MELAS: clinical, EEG, imaging, and biochemical factors. BMC Neurol. 2025 Feb 14;25(1):64. doi: 10.1186/s12883-025-04046-2[↩][↩]
- Scarpelli M, Zappini F, Filosto M, Russignan A, Tonin P, Tomelleri G. Mitochondrial Sensorineural Hearing Loss: A Retrospective Study and a Description of Cochlear Implantation in a MELAS Patient. Genet Res Int. 2012;2012:287432. doi: 10.1155/2012/287432[↩]
- Bhati RS, Sheridan BC, Mill MR, Selzman CH. Heart transplantation for progressive cardiomyopathy as a manifestation of MELAS syndrome. J Heart Lung Transplant. 2005 Dec;24(12):2286-9. doi: 10.1016/j.healun.2005.05.012[↩]
- Taivassalo T, Haller RG. Implications of exercise training in mtDNA defects–use it or lose it? Biochim Biophys Acta. 2004 Dec 6;1659(2-3):221-31. doi: 10.1016/j.bbabio.2004.09.007[↩]
- Koenig MK, Emrick L, Karaa A, Korson M, Scaglia F, Parikh S, Goldstein A. Recommendations for the Management of Strokelike Episodes in Patients With Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes. JAMA Neurol. 2016 May 1;73(5):591-4. doi: 10.1001/jamaneurol.2015.5072[↩]
- El-Hattab AW, Almannai M, Scaglia F. Arginine and citrulline for the treatment of MELAS syndrome. J Inborn Errors Metab Screen. 2017 Jan;5:10.1177/2326409817697399. doi: 10.1177/2326409817697399[↩]
- Lin CM, Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab Brain Dis. 2007 Mar;22(1):105-9. doi: 10.1007/s11011-006-9039-9. Epub 2007 Jan 17. Erratum in: Metab Brain Dis. 2007 Dec;22(3-4):425.[↩]
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006 Feb 14;66(3):324-30. doi: 10.1212/01.wnl.0000196641.05913.27[↩]
- Díaz-Lobato S, Gómez Mendieta MA, Moreno García MS, Mayoralas-Alises S, Arpa Gutierrez FJ. Two full-term pregnancies in a patient with mitochondrial myopathy and chronic ventilatory insufficiency. Respiration. 2005 Nov-Dec;72(6):654-6. doi: 10.1159/000089584[↩]
- Majamaa-Voltti KA, Winqvist S, Remes AM, Tolonen U, Pyhtinen J, Uimonen S, Kärppä M, Sorri M, Peuhkurinen K, Majamaa K. A 3-year clinical follow-up of adult patients with 3243A>G in mitochondrial DNA. Neurology. 2006 May 23;66(10):1470-5. doi: 10.1212/01.wnl.0000216136.61640.79[↩]




{kind=link}