Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a rare serious progressive (meaning it gets worse over time) condition characterized by abnormally high blood pressure (hypertension) in the pulmonary artery, the blood vessel that carries deoxygenated blood (venous blood) from the right side of your heart to your lungs, leading to increased resistance to blood flow and strain on the right side of the heart (see Figure 5 to 7 below) 1, 2, 3, 4, 5, 6, 7, 8. Blood (deoxygenated blood or venous blood) moves from your heart to your lungs through blood vessels called pulmonary arteries. The pulmonary artery carries blood from the right side of your heart to your lungs to pick up a fresh supply of oxygen. Once in your lungs, the deoxygenated blood (venous blood) travels through many small, thin blood vessels called capillaries. There, the blood picks up more oxygen and transfers carbon dioxide to the lungs—a process called gas exchange. The oxygen-rich blood passes from your lungs back to your heart through the pulmonary veins. If the small blood vessels in your lungs become narrowed, blocked or damaged, the blood does not flow through them as well. As a result, blood can’t flow through your lungs as well as it should. This can increase the blood pressure in the pulmonary arteries and cause pulmonary hypertension.
Pulmonary arterial hypertension (PAH) is one form of a broader serious condition known as pulmonary hypertension. In pulmonary hypertension most of the very small arteries throughout your lungs become narrowed in diameter, which increases the resistance to blood flow through your lungs, making it harder for blood to flow through them. To overcome the increased resistance, blood pressure increases in the pulmonary artery and in the right ventricle of your heart, which is the chamber that pumps blood into the pulmonary artery. Ultimately, the increased blood pressure can damage the right ventricle of your heart, which can lead to right-sided heart failure and potentially heart failure.
Primary pulmonary hypertension is classified in the World Health Organization’s (WHO) classification system as part of group 1. The WHO classification of pulmonary hypertension is based on the mechanism or underlying cause 4:
- Group 1: Pulmonary arterial hypertension (PAH) can be idiopathic (i.e., primary pulmonary hypertension) or due to congenital left to right intracardiac shunts, portal hypertension, persistent pulmonary hypertension of the newborn, collagen vascular diseases, HIV infection.
- Group 2: Pulmonary hypertension caused by left heart disease (pulmonary venous hypertension)
- Group 3: Pulmonary hypertension caused by lung diseases, hypoxia, or both
- Group 4: Chronic thromboembolic pulmonary hypertension and pulmonary hypertension caused by pulmonary artery obstructions
- Group 5: Pulmonary hypertension caused by unclear or multifactorial mechanisms.
For a person at rest, normal mean pulmonary artery pressure (mPAP) is typically considered to be less than 20 mmHg or between 8-20 millimeters of mercury (8-20 mmHg). In an individual with pulmonary hypertension, the resting mean pulmonary arterial blood pressure (mPAP) is 20 mmHg or higher measured during right heart catheterization, with pulmonary capillary wedge pressure (PAWP) less than 15 mm Hg and increased pulmonary vascular resistance (PVR) above 3 Wood units (240 dyne/second/cm5) at rest 9,10, 11, 12. Pulmonary artery wedge pressure (PAWP) also known as pulmonary artery occlusion pressure (PAOP) is a measurement of the pressure in the pulmonary arteries obtained by inflating a balloon-tipped catheter (often a Swan-Ganz catheter) that is “wedged” into a small branch of the pulmonary artery, pressure reading then reflects the pressure in the pulmonary veins and, therefore, the left atrium and it’s used to estimate left ventricular end-diastolic pressure (LVEDP), which is an indicator of the preload or filling volume of the left ventricle. Normally, pulmonary artery wedge pressure (PAWP) or pulmonary artery occlusion pressure (PAOP) is around 6 to 12 mmHg. An elevated pulmonary artery wedge pressure (PAWP) or pulmonary artery occlusion pressure (PAOP) greater than 18 mmHg in the context of normal oncotic pressure suggests left heart failure. Pulmonary vascular resistance (PVR) is the resistance against blood flow from the 4 pulmonary veins of the lung to the left atrium 13.
The term pulmonary artery hypertension (PAH) describes a subset of patients who also have the presence of pre-capillary hypertension, including an end-expiratory pulmonary artery wedge pressure (PAWP less than 15 mmHg) and a pulmonary vascular resistance (PVR) greater than 3 Woods units (240 dyne/second/cm5) at rest 1, 2, 3.
In the United States, about 1,000 new cases of pulmonary arterial hypertension (PAH) are diagnosed each year 14. Pulmonary arterial hypertension (PAH) is most common in women between the ages of 30-60 15. Pulmonary arterial hypertension (PAH) is 2 to 7 times times more likely to be diagnosed in females than males. Males over age 65 who develop pulmonary arterial hypertension (PAH) are more likely to have severe cases. Pulmonary arterial hypertension (PAH) can also affect infants. This condition is known as persistent pulmonary hypertension in the neonate (PPHN) or pulmonary hypertension in newborns.
The exact cause of pulmonary arterial hypertension (PAH) is unknown. It is unlike other forms of pulmonary hypertension, where high blood pressure in the lungs is caused by underlying heart or lung disease. Researchers believe that pulmonary arterial hypertension (PAH) occurs when there is injury to the cells that line the blood vessels of your lung, which over time results in this blood vessel disease. If the cause of this change is unknown it is referred to as idiopathic pulmonary arterial hypertension (IPAH) 16. If the change is believed to be caused by a genetic mutation it is called heritable pulmonary arterial hypertension or familial pulmonary arterial hypertension. Approximately 15-20% of pulmonary arterial hypertension (PAH) patients have heritable pulmonary arterial hypertension (familial pulmonary arterial hypertension). Heritable pulmonary arterial hypertension (HPAH) is a genetic condition caused by changes (mutations) in the BMPR2 gene most commonly, though other genes and pathways have recently been identified. In approximately 20% of families with PAH, doctors do not yet know the underlying gene variants despite clear evidence of a heritable pattern of disease. Other conditions that are associated with the development of pulmonary arterial hypertension (PAH) include: connective tissue disorders like scleroderma, systemic lupus erythematosus (SLE), critical congenital heart disease, or Down syndrome. Researchers have also identified nongenetic factors that increase the risk of developing pulmonary arterial hypertension. These include certain drugs used as appetite suppressants and several illegal drugs, such as cocaine and methamphetamine. Pulmonary arterial hypertension is also a rare complication of certain infectious diseases such as HIV and schistosomiasis or associated with other medical conditions like cirrhosis of the liver and congenital heart diseases.
Pulmonary arterial hypertension (PAH) causes and subtypes 17:
- Idiopathic pulmonary arterial hypertension (IPAH)
- Non-responders at vasoreactivity testing
- Acute responders at vasoreactivity testing
- Heritable pulmonary arterial hypertension (HPAH) also called familial pulmonary arterial hypertension (FPAH)
- Pulmonary arterial hypertension associated with drugs and toxins 12
- Aminorex
- Fenfluramine
- Dexfenfluramine
- Benfluorex
- Methamphetamines
- Dasatinib
- Toxic rapeseed oil
- Cocaine
- Phenylpropanolamine
- L-tryptophan
- St John’s wort
- Amphetamines
- Interferon-α and -β
- Alkylating agents
- Bosutinib
- Direct-acting antiviral agents against hepatitis C virus
- Leflunomide
- Indirubin (Chinese herb Qing-Dai)
- Pulmonary arterial hypertension associated with:
- Connective tissue disease
- HIV infection
- Portal hypertension
- Congenital heart disease
- Schistosomiasis
- Pulmonary arterial hypertension (PAH) with features of venous/capillary involvement e.g., pulmonary capillary hemangiomatosis or pulmonary veno-occlusive disease
- Persistent pulmonary hypertension of the newborn (PPHN)
Pulmonary arterial hypertension (PAH) signs and symptoms occur when the increased blood pressure in the pulmonary artery cannot fully overcome the increased resistance to blood flow. As a result, the flow of oxygenated blood from your lungs to the rest of the body is insufficient. Shortness of breath during physical activity (exertional dyspnea) and fainting spells are the most common symptoms of pulmonary arterial hypertension (PAH). Some people with pulmonary arterial hypertension may experience additional symptoms, particularly as the condition worsens. Other symptoms include dizziness, swelling (edema) of the ankles or legs, fatigue, chest pain, and a rapid heart rate. If left untreated, pulmonary arterial hypertension (PAH) can lead to serious complications such as right heart failure, arrhythmias, and blood clots.
Pulmonary arterial hypertension (PAH) diagnosis involves a combination of medical history, physical exam, and tests such as echocardiogram and cardiac catheterization. Your doctor may also refer you to a pulmonologist (lung specialist) or cardiologist (heart specialist). These specialists will run specific tests to check your heart and lung function. They’ll determine what form of pulmonary hypertension you have (PAH or another form). They’ll also evaluate how far your condition has progressed.
There’s no cure for pulmonary arterial hypertension (PAH). Treatment options vary from person to person and depends on the cause, the type and severity of the pulmonary arterial hypertension. If you are newly diagnosed with pulmonary arterial hypertension (PAH), you should be referred to an accredited pulmonary hypertension care center for thorough evaluation. Because pulmonary arterial hypertension (PAH) is such a rare disease, it is extremely valuable to see a specialist at an accredited center to ensure you are getting the most up-to-date treatment options.
Figure 1. Pulmonary arterial hypertension
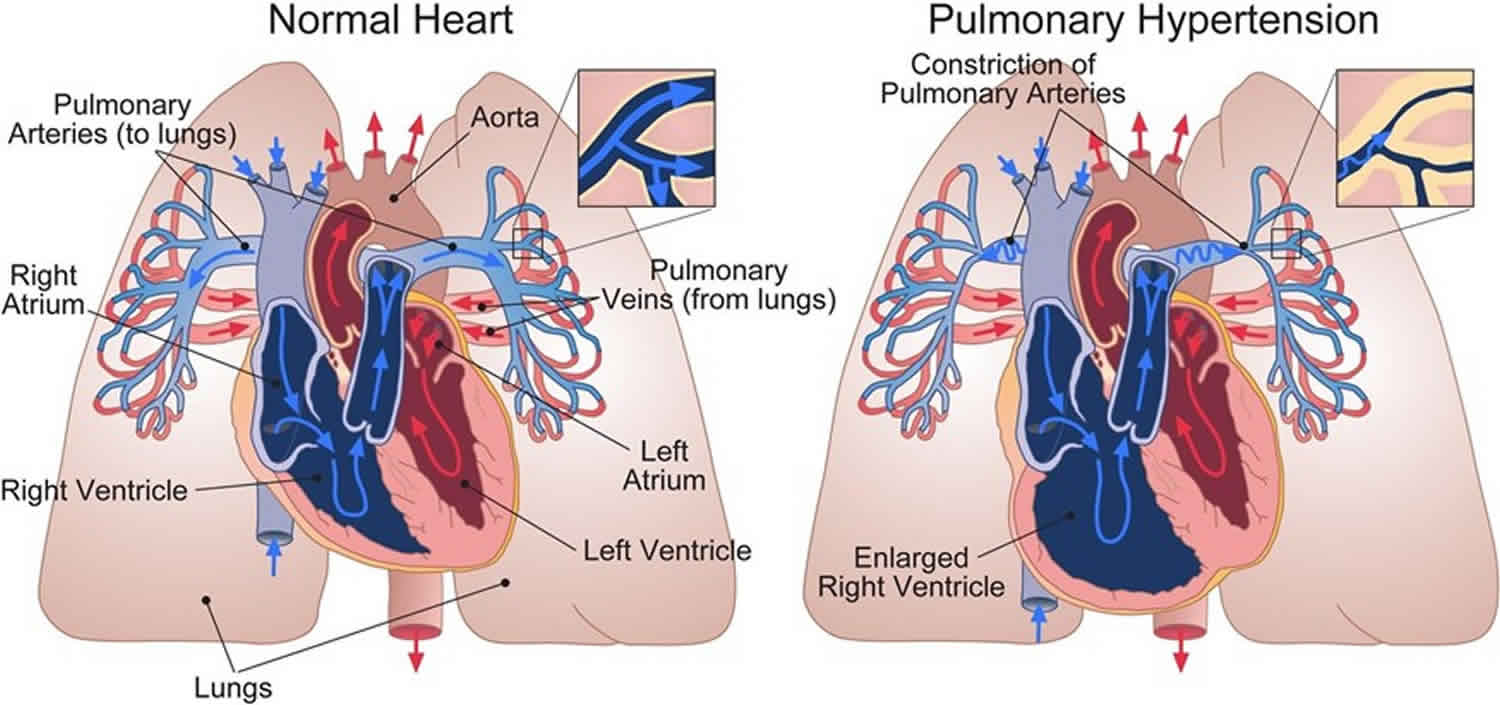
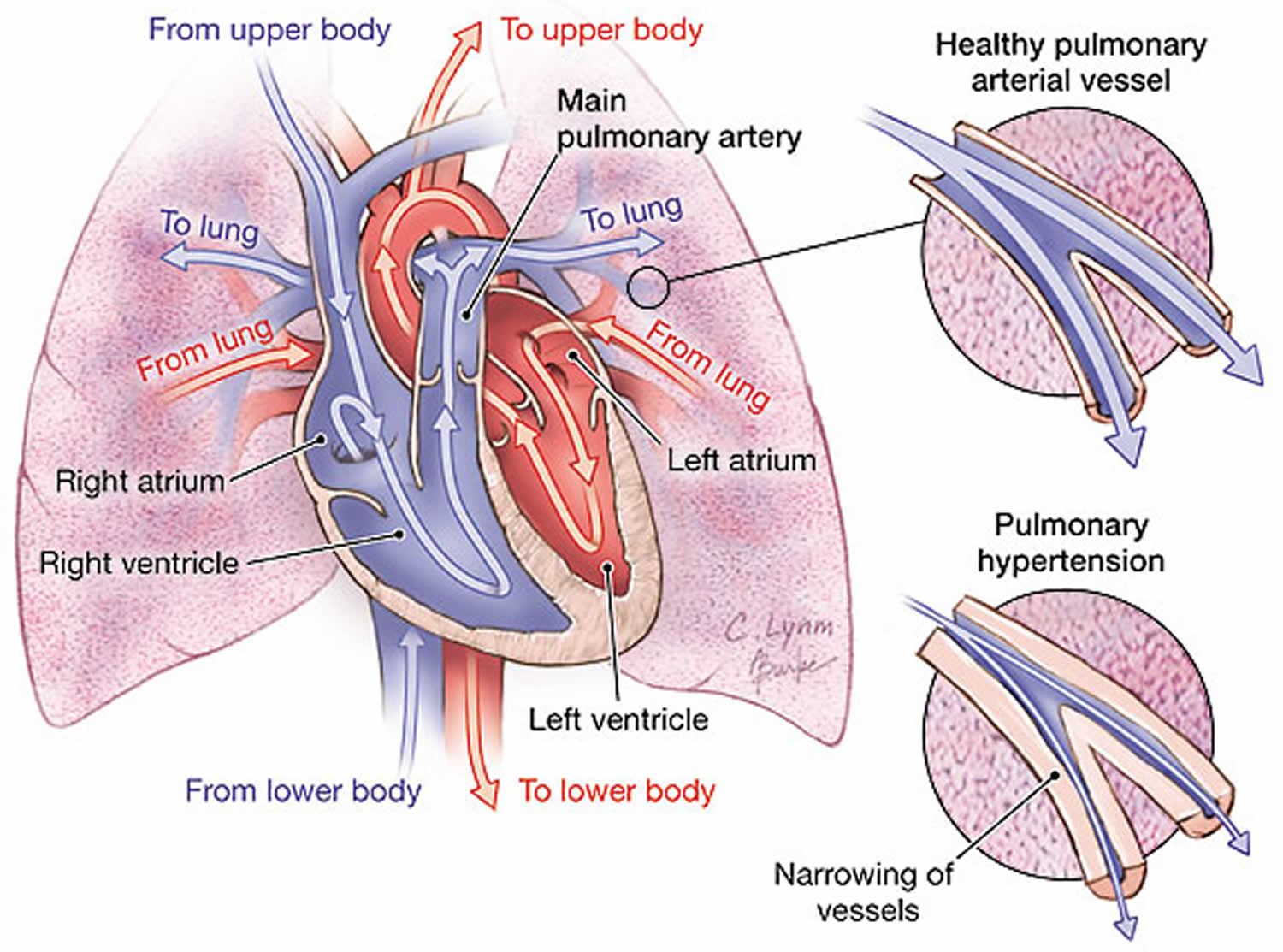
Figure 2. Pulmonary artery anatomy
Figure 3. Pulmonary arterial hypertension pathophysiology and current therapeutic targets
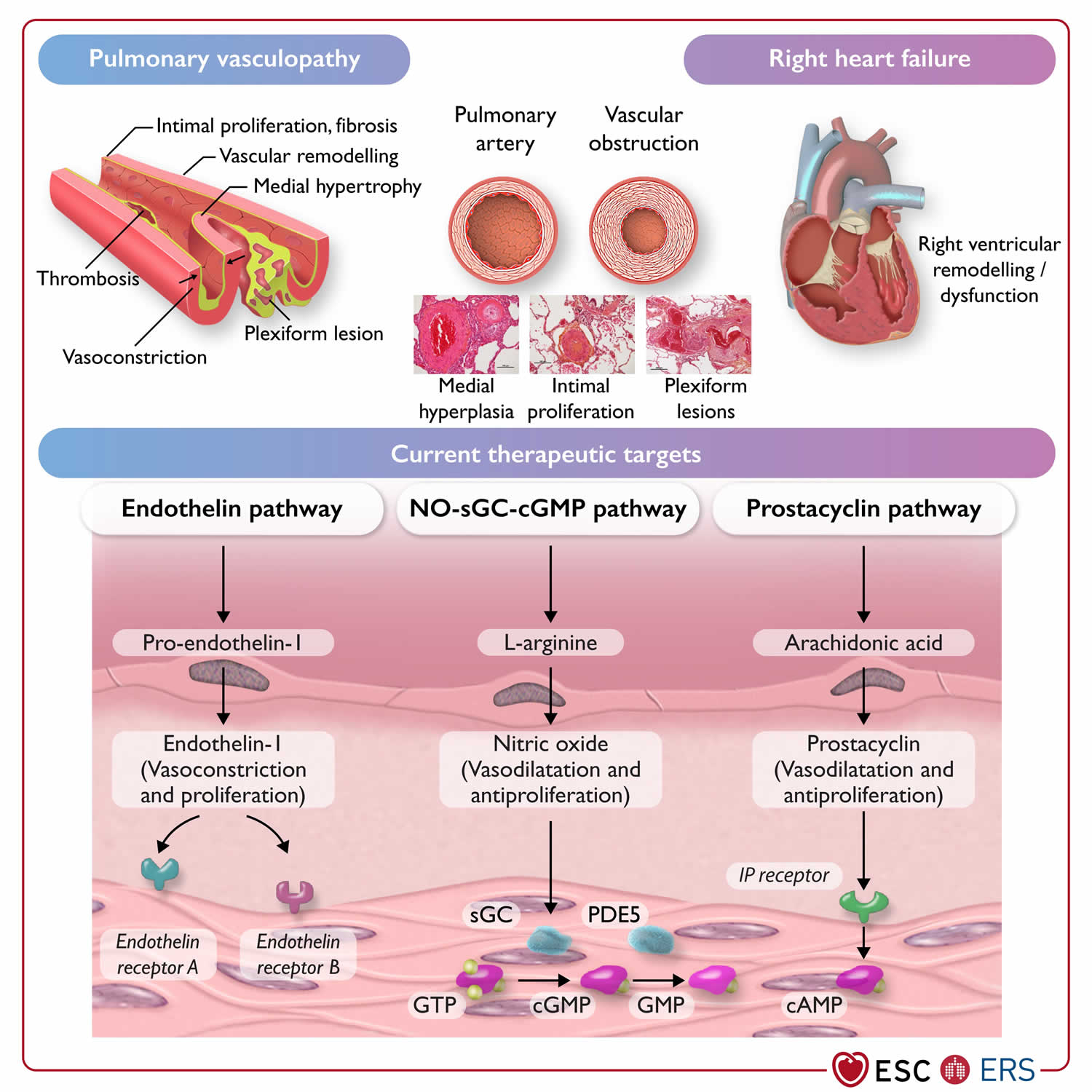
Footnotes: Pathophysiology and current therapeutic targets of pulmonary arterial hypertension (group 1).
Abbreviations: cAMP, cyclic adenosine monophosphate; (c)GMP, (cyclic) guanosine monophosphate; GTP, guanosine-5′-triphosphate; IP receptor, prostacyclin I2 receptor; NO, nitric oxide; PDE5, phosphodiesterase 5; sGC, soluble guanylate cyclase.
[Source 4 ]Figure 4. Pulmonary hypertension diagnostic algorithm
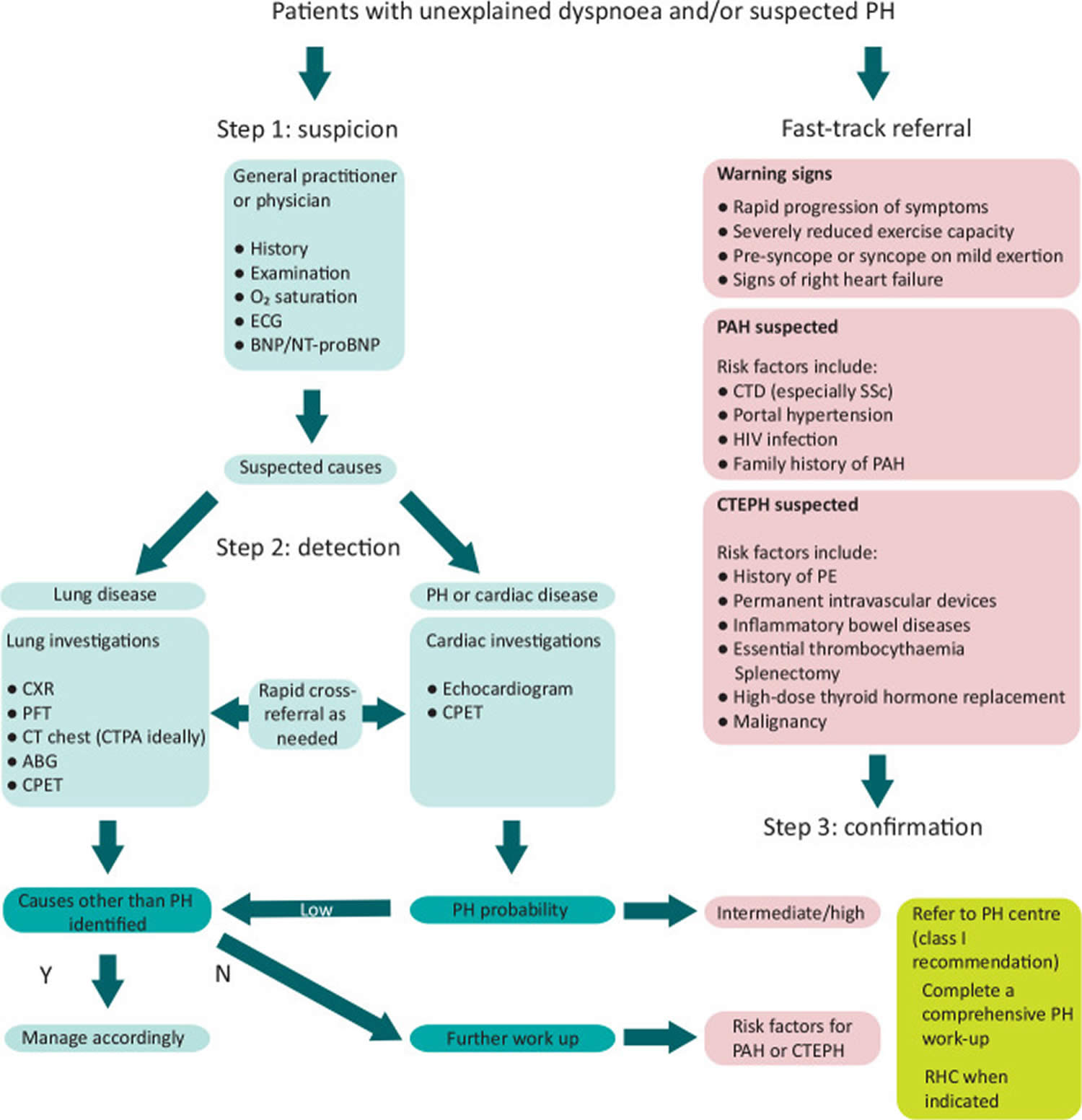
Footnotes: Diagnostic algorithm for patients with unexplained dyspnea and/or suspected pulmonary hypertension. Adapted from the 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension 4.
Abbreviations: ABG = arterial blood gas; BNP = brain natriuretic peptide; CPET = cardiopulmonary exercise testing; CT = computed tomography; CTEPH = chronic thromboembolic pulmonary hypertension; ECG = electrocardiogram; HIV = human immunodeficiency virus; N = no; NT-proBNP = N-terminal pro-brain natriuretic peptide; PAH = pulmonary arterial hypertension; PE = pulmonary embolism; PFT = pulmonary function tests; PH = pulmonary hypertension; Y = yes.
[Source 17 ]See your doctor if you’re having problems with:
- A fast heart rate (120 beats per minute).
- A respiratory infection or cough that’s getting worse.
- Constantly feeling dizzy or lightheaded.
- Episodes of chest pain or discomfort with physical activity.
- Extreme fatigue or decreased ability to do your normal activities.
- Nausea or lack of appetite.
- Restlessness or confusion.
- Shortness of breath that’s gotten worse, especially if you wake up feeling short of breath.
- Swelling in your ankles, legs or stomach that’s gotten worse.
- Trouble breathing with regular activities or at rest.
- Weight gain (2 pounds in one day or 5 pounds in one week).
When should I go to the emergency room?
Go to the emergency department (ER) or call your local emergency number if you have:
- A fast heart rate (120-150 beats per minute) that won’t go down.
- Fainting spells with loss of consciousness.
- Complications with your IV or infusion pump. These include infection, catheter displacement, solution leak, bleeding and IV pump malfunction.
- Shortness of breath that doesn’t go away when you rest.
- Sudden and severe chest pain.
- Sudden and severe headache.
- Sudden weakness or paralysis in your arms or legs.
How common is pulmonary arterial hypertension?
Pulmonary arterial hypertension (PAH) isn’t as common as other forms of pulmonary hypertension, including those caused by underlying heart or lung disease. Each year, about 500 to 1,000 people are diagnosed with pulmonary arterial hypertension (PAH) in the U.S 15. In Western countries, about 25 per 1 million people are living with pulmonary arterial hypertension (PAH). In a U.S. registry of 2039 patients with pulmonary artery hypertension (PAH), the average age was 51.7 (plus or minus 14.5 years) with a female-to-male ratio of 3.9 to 1. Of those patients, 46.6% (950) were classified as idiopathic PAH (IPAH). Estimates are that idiopathic PAH (IPAH) affects 1 in 1 million, usually young females who are otherwise normal. The median survival, if left untreated, is 2.8 years 18, 19.
How does pulmonary arterial hypertension affect my body?
Pulmonary arterial hypertension strains the right side of your heart, which pumps oxygen-poor blood to your lungs. This strain can lead to right-sided heart failure. Furthermore, pulmonary arterial hypertension (PAH) slows down blood flow between your heart and lungs. This means less blood can enter your lungs to gain fresh oxygen. As a result, blood flow to the rest of your body also slows down. So, your organs and tissues can’t get enough oxygen. Without treatment, pulmonary arterial hypertension (PAH) can be fatal.
How serious is pulmonary arterial hypertension?
Pulmonary arterial hypertension (PAH) is a serious condition that can be life-threatening. An early diagnosis and swift treatment can help you live longer and have a better quality of life.
Can pulmonary arterial hypertension be reversed?
Currently, medications can slow down pulmonary arterial hypertension (PAH) progression but not reverse the damage already done. However, researchers are working on promising new medications that could help reverse pulmonary arterial hypertension (PAH). Such medications would repair damage to the endothelial cells that line your pulmonary arteries.
Talk with your doctor to learn more about the latest research and clinical trials for pulmonary arterial hypertension (PAH) therapies.
Who does pulmonary arterial hypertension affect?
Pulmonary arterial hypertension (PAH) can affect adults at any age. It’s more common among females, who are usually diagnosed between the ages of 30 and 60. Males over age 65 who develop pulmonary arterial hypertension (PAH) are more likely to have severe cases. Pulmonary arterial hypertension (PAH) can also affect infants. This condition is known as persistent pulmonary hypertension in the neonate (PPHN) or pulmonary hypertension in newborns.
Pregnancy and pulmonary arterial hypertension
Historically, pregnancy in women with pulmonary arterial hypertension (PAH) and other forms of severe pulmonary hypertension has been associated with maternal mortality rates of up to 56% and neonatal mortality rates of up to 13% 20. With improved treatment of pulmonary arterial hypertension (PAH) and new approaches to managing women during pregnancy and the peri-partum period, maternal mortality has declined but remains high, ranging 11–25% 21, 22, 23, 24, 25. For these reasons, previous European Society of Cardiology and European Respiratory Society Guidelines for the diagnosis and treatment of pulmonary hypertension have recommended that patients with pulmonary arterial hypertension should avoid pregnancy 26, 27. However, there are reports of favourable pregnancy outcomes in women with pulmonary hypertension, including, but not limited to, women with idiopathic pulmonary arterial hypertension (IPAH) who respond to calcium channel blocker therapy 23, 24, 28, 29. Nonetheless, pregnancy remains associated with unforeseeable risks, and may accelerate pulmonary hypertension progression 30. Women with pulmonary hypertension can deteriorate at any time during or after pregnancy. Therefore, physicians have a responsibility to inform patients about the risks of pregnancy, so that women and their families can make informed decisions.
Women with poorly controlled disease, indicated by an intermediate- or high-risk profile and signs of right ventricular dysfunction, are at high risk of adverse outcomes; in the event of pregnancy, they should be carefully counseled and early termination should be advised 4. For patients with well-controlled disease, a low-risk profile, and normal or near-normal resting hemodynamics who consider becoming pregnant, individual counselling and shared decision-making are recommended 4. In such cases, alternatives such as adoption and surrogacy may also be explored. Pre-conception genetic counselling should also be considered in heritable pulmonary arterial hypertension.
Women with pulmonary hypertension who become pregnant or present during pregnancy with newly diagnosed PAH should be treated, whenever possible, in centers with a multidisciplinary team experienced in managing pulmonary hypertension in pregnancy 4. If pregnancy is continued, PAH therapy may have to be adjusted. It is recommended to stop endothelin receptor antagonists (ERAs), riociguat, and selexipag because of potential or unknown teratogenicity 31. Despite limited evidence, calcium channel blockers, Phosphodiesterase 5 inhibitors, and inhaled/i.v./subcutaneous (s.c.) prostacyclin analogues are considered safe during pregnancy 28, 32.
Pregnancy in pulmonary hypertension is a very sensitive topic and requires empathic communication. Psychological support should be offered whenever needed.
Birth control
Women with pulmonary hypertension of childbearing potential should be provided with clear contraceptive advice, considering the individual needs of the woman but recognizing that the implications of contraceptive failure are significant in pulmonary hypertension. With appropriate use, many forms of contraception, including oral contraceptives, are highly effective. In patients treated with bosentan, reduced efficacy of hormonal contraceptives should be carefully considered 33. Using hormonal implants or an intrauterine device (IUD) are alternative options with low failure rates. Surgical sterilization may be considered but is associated with peri-operative risks. Emergency post-coital hormonal contraception is safe in pulmonary hypertension.
How your Heart Works
Your heart is a muscular organ that pumps blood to your body. Your heart is at the center of your circulatory system. The circulatory system consists of a network of blood vessels, such as arteries, veins, and capillaries. These blood vessels carry blood to and from all areas of your body.
An electrical system controls your heart and uses electrical signals to contract the heart’s walls. When the heart walls contract, blood is pumped into your circulatory system. Inlet and outlet valves in your heart chambers ensure that blood flows in the right direction.
Your heart is vital to your health and nearly everything that goes on in your body. Without your heart’s pumping action, blood can’t move throughout your body.
Your blood carries the oxygen and nutrients that your organs need to work well. Blood also carries carbon dioxide (a waste product) to your lungs so you can breathe it out.
A healthy heart supplies your body with the right amount of blood and oxygen at the rate needed to work well. If disease or injury weakens your heart, your body’s organs won’t receive enough blood to work normally.
Your heart has 2 sides, separated by an inner wall called the septum. The right side of the heart pumps blood to your lungs to pick up oxygen. The left side of your heart receives the oxygen-rich blood from the lungs and pumps it to the body.
Figure 5. The anatomy of human heart
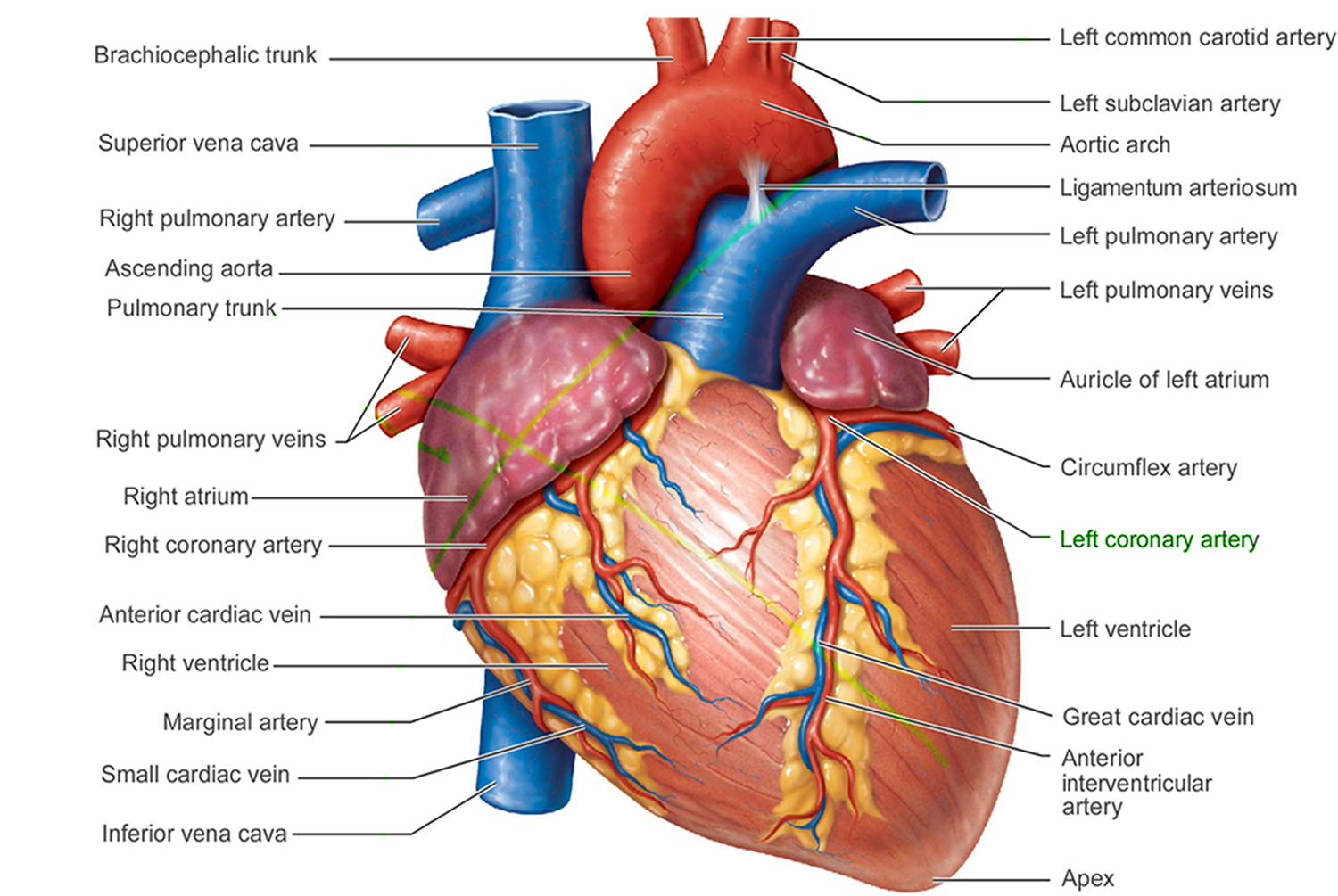
Figure 6. The anatomy of the heart chambers
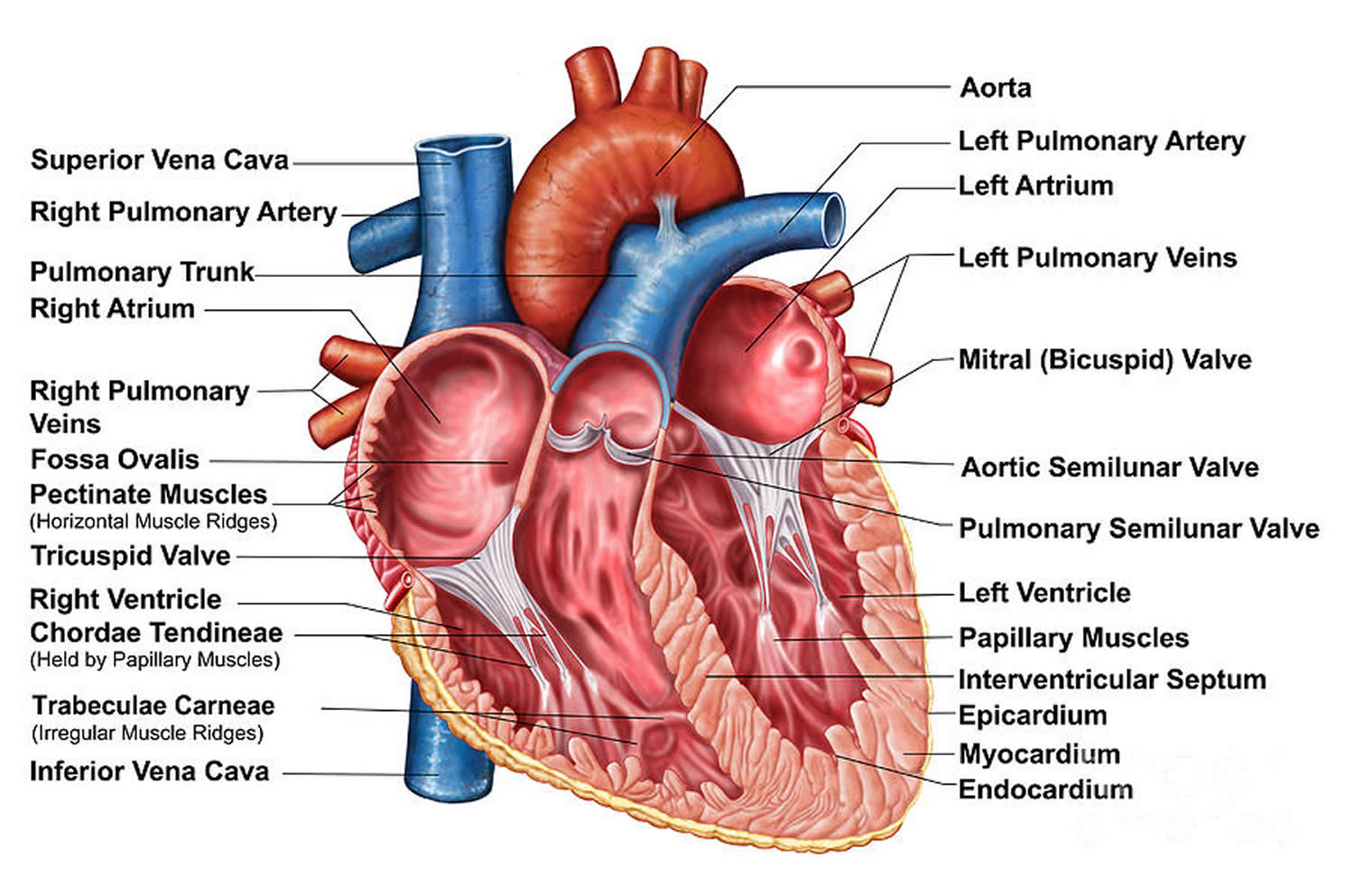
Figure 7. Normal heart blood flow
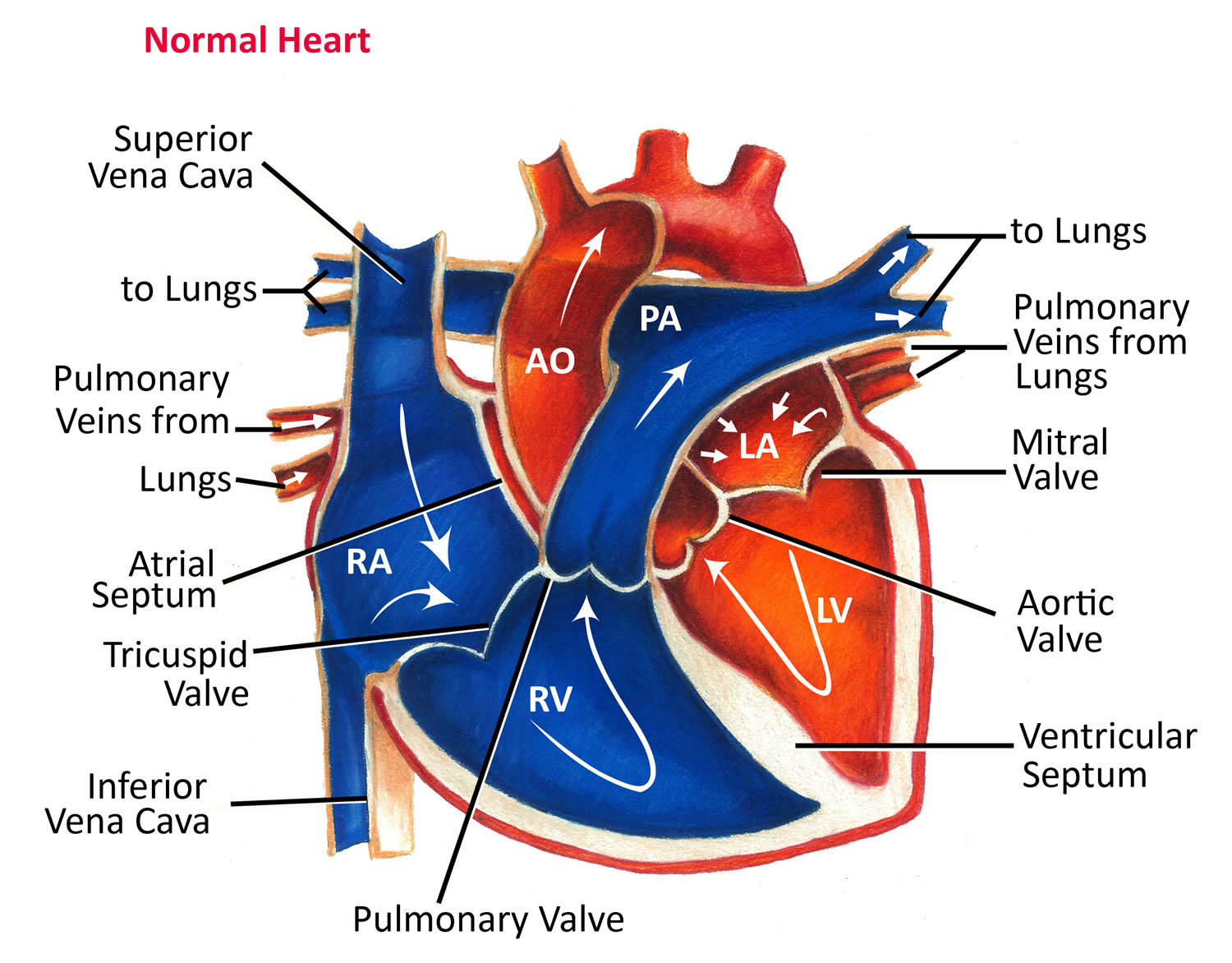
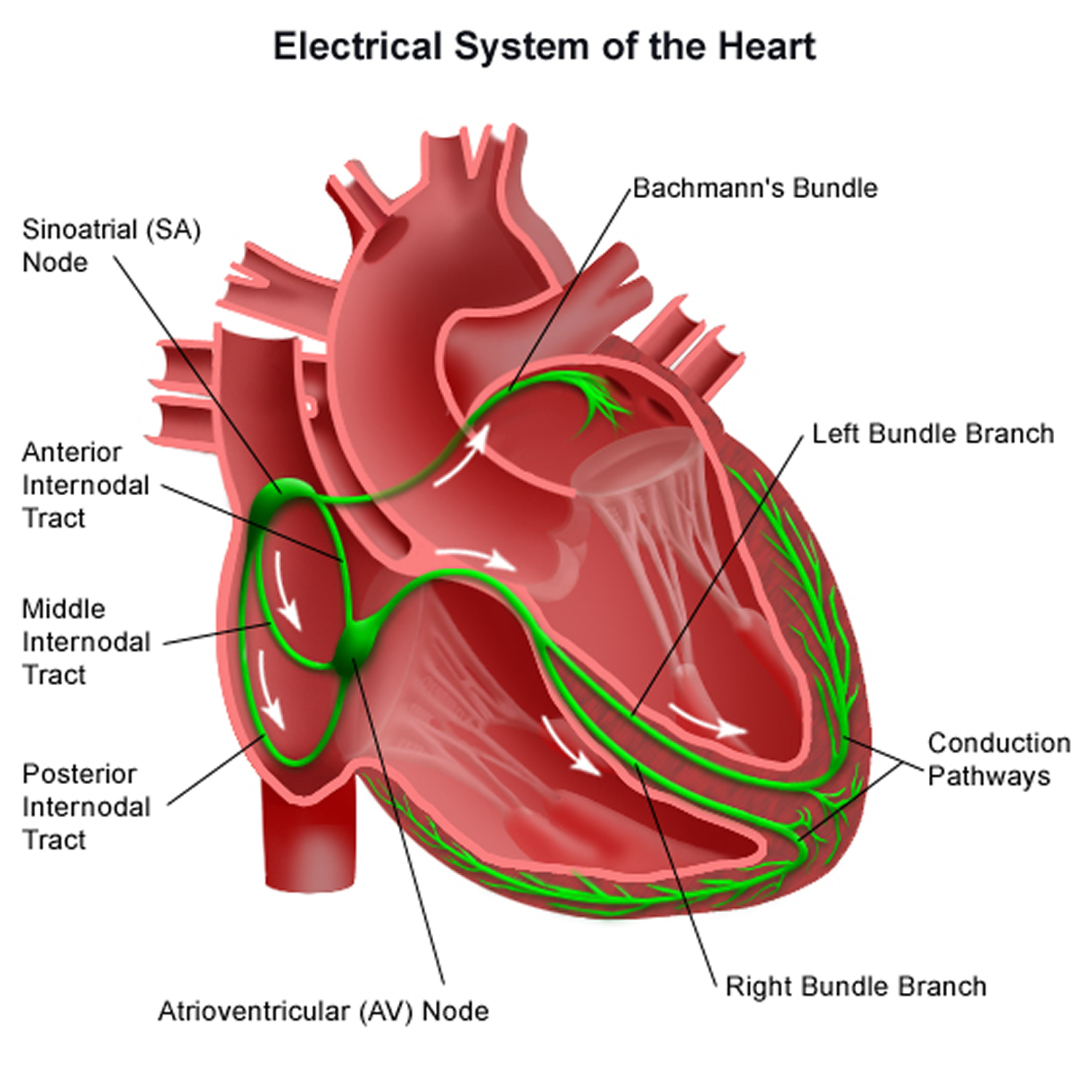
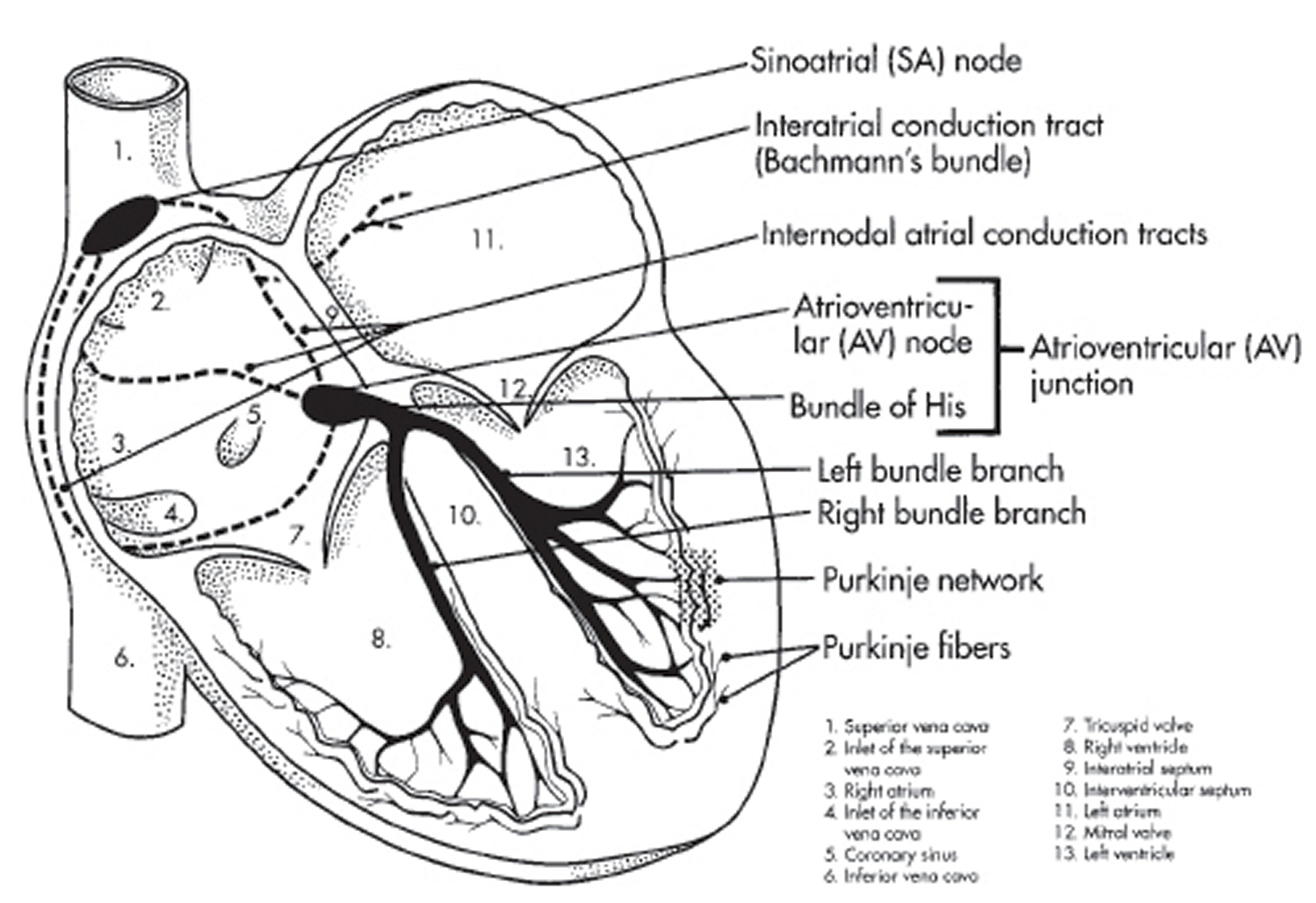
Figure 8. The heart’s electrical system
Heart Chambers
Your heart has 4 chambers 34, 2 on the right and 2 on the left:
- 2 upper chambers are called atrium (two is called an atria). The atria collect blood as it flows into your heart.
- 2 lower chambers are called ventricles.
- Right ventricle (RV) pumps Deoxygenated blood out of your heart to your lungs. Deoxygenated blood, also known as venous blood, is blood that has a lower oxygen concentration and a higher concentration of carbon dioxide than oxygenated blood.
- Left ventricle (LV) pumps Oxygenated blood out of your heart to other parts of your body. Oxygenated blood, also known as arterial blood, is blood rich in oxygen, typically bright red, that is pumped from the left ventricle of your heart to your body through arteries (aorta) after picking up oxygen in your lungs.
Your heart also has 4 valves that open and close to let blood flow from the atria to the ventricles and from the ventricles into the two large arteries connected to the heart in only one direction when the heart contracts (beats). The four heart valves are:
- Tricuspid valve, located between the right atrium and right ventricle
- Pulmonary or pulmonic valve, between the right ventricle and the pulmonary artery. The pulmonary artery carries blood from your heart to your lungs.
- Mitral valve, between the left atrium and left ventricle
- Aortic valve, between the left ventricle and the aorta. This aorta carries blood from the heart to the body.
Each valve has a set of flaps also called leaflets or cusps. The mitral valve has two flaps; the others have three. Valves are like doors that open and close. They open to allow blood to flow through to the next chamber or to one of the arteries. Then they shut to keep blood from flowing backward. Blood flow occurs only when there’s a difference in pressure across the valves, which causes them to open. Under normal conditions, the valves permit blood to flow in only one direction.
Your heart 4 chambers and 4 valves and is connected to various blood vessels. Veins are blood vessels that carry blood from the body to your heart. Arteries are blood vessels that carry blood away from your heart to your body.
Your heart pumps blood to your lungs and to all the body’s tissues by a sequence of highly organized contractions of the four chambers. For your heart to function properly, the four chambers must beat in an organized way.
When your heart’s valves open and close, they make a “lub-DUB” sound that a doctor can hear using a stethoscope 35.
- The First heart sound (S1) — the “lub” —is made by the mitral and tricuspid valves closing at the beginning of systole. Systole is when the ventricles contract, or squeeze, and pump blood out of the heart.
- The Second heart sound (S2) — the “DUB” —is made by the aortic and pulmonary valves closing at the beginning of diastole. Diastole is when the ventricles relax and fill with blood pumped into them by the atria.
Arteries
The arteries are major blood vessels connected to your heart.
- Pulmonary artery carries blood from the right side of the heart to the lungs to pick up a fresh supply of oxygen.
- Aorta is the main artery that carries oxygen-rich blood from the left side of the heart to the body.
- Coronary arteries are the other important arteries attached to the heart. They carry oxygen-rich blood from the aorta to the heart muscle, which must have its own blood supply to function.
Veins
The veins also are major blood vessels connected to your heart.
- Pulmonary veins carry oxygen-rich blood from the lungs to the left side of the heart so it can be pumped to the body.
- Superior vena cava (SVC) and inferior vena cava (IVC) are large veins that carry oxygen-poor blood (deoxygenated blood or venous blood) from the body back to the heart.
Blood Flow
The Right Side of Your Heart
In figure 5 above, the superior vena cava (SVC) and inferior vena cava (IVC) are shown in blue to the left of the heart muscle as you look at the picture. The superior vena cava (SVC) and inferior vena cava (IVC) are the largest veins in your body.
After your body’s organs and tissues have used the oxygen in your blood, the vena cavae carry the oxygen-poor blood (deoxygenated blood or venous blood) back to the right atrium of your heart.
- The superior vena cava (SVC) carries oxygen-poor blood from the upper parts of your body, including your head, chest, arms, and neck.
- The inferior vena cava (IVC) carries oxygen-poor blood from the lower parts of your body from the abdomen and lower extremities back to the right side of your heart for oxygenation.
The oxygen-poor blood (deoxygenated blood or venous blood) from the vena cavae flows into your heart’s right atrium. From the right atrium, blood is pumped into the right ventricle. And then from the right ventricle, blood is pumped to your lungs through the pulmonary arteries (shown in blue in the center of figure 5).
Once in the lungs, the blood travels through many small, thin blood vessels called capillaries. There, the blood picks up more oxygen and transfers carbon dioxide to the lungs—a process called gas exchange.
The oxygen-rich blood passes from your lungs back to your heart through the pulmonary veins (shown in red to the left of the right atrium in figure 5).
The Left Side of Your Heart
Oxygen-rich blood from your lungs passes through the pulmonary veins (shown in red to the right of the left atrium in figure 5 above). The blood enters the left atrium and is pumped into the left ventricle.
From the left ventricle, the oxygen-rich blood is pumped to the rest of your body through the aorta. The aorta is the main artery that carries oxygen-rich blood to your body.
Like all of your organs, your heart needs oxygen-rich blood. As blood is pumped out of your heart’s left ventricle, some of it flows into the coronary arteries (shown in red in figure 5).
Your coronary arteries are located on your heart’s surface at the beginning of the aorta. They carry oxygen-rich blood to all parts of your heart.
For the heart to work well, your blood must flow in only one direction. Your heart’s valves make this possible. Both of your heart’s ventricles have an “in” (inlet) valve from the atria and an “out” (outlet) valve leading to your arteries.
Healthy valves open and close in exact coordination with the pumping action of your heart’s atria and ventricles. Each valve has a set of flaps called leaflets or cusps that seal or open the valve. This allows blood to pass through the chambers and into your arteries without backing up or flowing backward.
Heart’s Electrical System
To understand arrhythmias, it helps to understand the heart’s internal electrical system. The heart’s electrical system controls the rate and rhythm of the heartbeat.
With each heartbeat, an electrical signal spreads from the top of the heart to the bottom. As the signal travels, it causes the heart to contract and pump blood.
Your heart’s electrical system controls all the events that occur when your heart pumps blood 36. The electrical system also is called the cardiac conduction system. If you’ve ever seen the heart test called an EKG (electrocardiogram), you’ve seen a graphical picture of the heart’s electrical activity.
Your heart’s electrical system is made up of three main parts:
- Sinoatrial (SA) node, located in the right atrium of your heart
- Atrioventricular (AV) node, located on the interatrial septum close to the tricuspid valve
- His-Purkinje system, located along the walls of your heart’s ventricles
A heartbeat is a complex series of events. These events take place inside and around your heart. A heartbeat is a single cycle in which your heart’s chambers relax and contract to pump blood. This cycle includes the opening and closing of the inlet and outlet valves of the right and left ventricles of your heart.
Each heartbeat has two basic parts: diastole and systole. During diastole, the atria and ventricles of your heart relax and begin to fill with blood.
At the end of diastole, your heart’s atria contract (atrial systole) and pump blood into the ventricles. The atria then begin to relax. Your heart’s ventricles then contract (ventricular systole), pumping blood out of your heart.
Each electrical signal begins in a group of cells called the sinus node or sinoatrial (SA) node. The SA node is located in the heart’s upper right chamber, the right atrium. In a healthy adult heart at rest, the SA node fires off an electrical signal to begin a new heartbeat 60 to 100 times a minute. In a normal, healthy heart, each beat begins with a signal from the SA node. This is why the SA node sometimes is called your heart’s natural pacemaker. Your pulse, or heart rate, is the number of signals the SA node produces per minute.
The signal is generated as the vena cavae fill your heart’s right atrium with blood from other parts of your body. The signal spreads across the cells of your heart’s right and left atria.
From the SA node, the electrical signal travels through special pathways in the right and left atria. This causes the atria to contract and pump blood through the open valves from the atria into heart’s two lower chambers, the ventricles.
The electrical signal then moves down to a group of cells called the atrioventricular (AV) node, located between the atria and the ventricles. Here, the signal slows down just a little, allowing your heart’s right and left ventricles time to finish filling with blood.
The electrical signal then leaves the AV node and travels along a pathway called the bundle of His. This pathway divides into a right bundle branch and a left bundle branch. The signal goes down these branches to the ventricles, causing them to contract and pump blood to the lungs and the rest of the body.
From the bundle of His, the signal fibers divide into left and right bundle branches through the Purkinje fibers. These fibers connect directly to the cells in the walls of your heart’s left and right ventricles.
The signal spreads across the cells of your ventricle walls, and both ventricles contract. However, this doesn’t happen at exactly the same moment.
The left ventricle contracts an instant before the right ventricle. This pushes blood through the pulmonary valve (for the right ventricle) to your lungs, and through the aortic valve (for the left ventricle) to the rest of your body.
As the signal passes, the walls of the ventricles relax and await the next signal.
This process continues over and over as the atria refill with blood and more electrical signals come from the SA node.
A problem with any part of this process can cause an arrhythmia. For example, in atrial fibrillation, a common type of arrhythmia, electrical signals travel through the atria in a fast and disorganized way. This causes the atria to quiver instead of contract.
Pulmonary hypertension types and causes
| Classification* | Epidemiology | Hemodynamic characteristics† | Treatment |
|---|---|---|---|
| Group 1‡ | |||
|
|
| |
| Group 2‡ | |||
|
|
| |
| Group 3‡ | |||
|
|
| |
| Group 4‡ | |||
|
|
| |
| Group 5‡ | |||
|
|
|
|
Footnotes: World Health Organization (WHO) Pulmonary hypertension classification, epidemiology, hemodynamic characteristics, and treatments.
‡ Pulmonary hypertension is clinically divided into 5 groups:
- Group 1: Pulmonary arterial hypertension (PAH)
- Group 2: Pulmonary hypertension caused by left heart disease
- Group 3: Pulmonary hypertension caused by lung diseases, hypoxia, or both
- Group 4: Chronic thromboembolic pulmonary hypertension and pulmonary hypertension caused by pulmonary artery obstructions
- Group 5: Pulmonary hypertension caused by unclear or multifactorial mechanisms.
* According to the Sixth World Symposium on Pulmonary Hypertension.
† Defined by Sixth World Symposium on Pulmonary Hypertension, 2018. The 2022 European Respiratory Society-European Society of Cardiology guidelines suggest a cut-off for pulmonary vascular resistance of 2 Woods units.
Abbreviations: COPD = chronic obstructive pulmonary disease; COPD-PH = pulmonary hypertension related to COPD; ILD-PH = pulmonary hypertension related to interstitial lung disease; LVEF = left ventricular ejection fraction; mPAP = mean pulmonary arterial pressure; OSA = obstructive sleep apnea; PaCO2 = partial pressure of carbon dioxide in arterial blood; PAH = pulmonary arterial hypertension; PAWP = pulmonary arterial wedge pressure; PH = pulmonary hypertension; PVR = pulmonary vascular resistance; SpO2 = arterial oxygen saturation measured by pulse oximeter.
[Source 49 ]Table 1. Hemodynamic definitions of pulmonary hypertension
| Definitions | Characteristics | WHO Clinical Groups# |
|---|---|---|
| Pulmonary hypertension (PH) | Mean pulmonary arterial pressure (mPAP) >20 mmHg | |
| Pre-capillary pulmonary hypertension | Mean pulmonary arterial pressure (mPAP) >20 mmHg | 1, 3, 4 and 5 |
| Pulmonary arterial wedge pressure (PAWP) ≤15 mmHg | ||
| Pulmonary vascular resistance (PVR) ≥3 Wood Units | ||
| Isolated post-capillary pulmonary hypertension (IpcPH) | mPAP >20 mmHg | 2 and 5 |
| PAWP >15 mmHg | ||
| PVR <3 WU | ||
| Combined pre- and post-capillary pulmonary hypertension (CpcPH) | mPAP >20 mmHg | 2 and 5 |
| PAWP >15 mmHg | ||
| PVR ≥3 WU | ||
| Exercise pulmonary hypertension *** | mPAP/cardiac output slope between rest and exercise >3 mmHg/L/min |
Abbreviations: mPAP = mean pulmonary arterial pressure; PAWP = pulmonary arterial wedge pressure; PVR = pulmonary vascular resistance; WU = Wood Units. #: Group 1 = pulmonary arterial hypertension (PAH); Group 2 = pulmonary hypertension due to left heart disease; Group 3 = pulmonary hypertension due to lung diseases and/or hypoxia; Group 4 = pulmonary hypertension due to pulmonary artery obstructions; Group 5 = pulmonary hypertension with unclear and/or multifactorial mechanisms.
Footnotes: *** Exercise pulmonary hypertension, defined by an mPAP/cardiac output (CO) slope >3 mmHg/L/min between rest and exercise, has been re-introduced 50. The mPAP/cardiac output (CO) slope is strongly age dependent and its upper limit of normal ranges from 1.6–3.3 mmHg/L/min in the supine position 50. An mPAP/cardiac output (CO) slope >3 mmHg/L/min is not physiological in subjects aged <60 years and may rarely be present in healthy subjects aged >60 years 50. A pathological increase in pulmonary pressure during exercise is associated with impaired prognosis in patients with exercise dyspnea 51 and in several cardiovascular conditions 52, 53, 54, 55. Although an increased mPAP/CO slope defines an abnormal hemodynamic response to exercise, it does not allow for differentiation between pre- and post-capillary causes. The PAWP/CO slope with a threshold >2 mmHg/L/min may best differentiate between pre- and post-capillary causes of exercise pulmonary hypertension 56, 57.
[Source 58 ]Pulmonary arterial hypertension causes
Pulmonary arterial hypertension (PAH) causes include:
- Unknown cause, called idiopathic pulmonary arterial hypertension (IPAH).
- Changes in a gene passed down through families, called heritable pulmonary arterial hypertension (HPAH).
- Use of some medicines or illegal drugs, including methamphetamine and cocaine.
- Heart problems present at birth, called a congenital heart disease or congenital heart defect.
- Other health conditions, including scleroderma, systemic lupus erythematosus (SLE) and chronic liver disease such as cirrhosis.
- Certain infectious diseases such as HIV and schistosomiasis
The exact cause of pulmonary arterial hypertension (PAH) is unknown. It is unlike other forms of pulmonary hypertension, where high blood pressure in the lungs is caused by underlying heart or lung disease. Researchers believe that pulmonary arterial hypertension (PAH) occurs when there is injury to the cells that line the blood vessels of your lung, which over time results in this blood vessel disease. If the cause of this change is unknown it is referred to as idiopathic pulmonary arterial hypertension (PAH). If the change is believed to be caused by a genetic mutation it is called heritable pulmonary arterial hypertension or familial pulmonary arterial hypertension. Approximately 15-20% of pulmonary arterial hypertension (PAH) patients have heritable pulmonary arterial hypertension (familial pulmonary arterial hypertension). Other conditions that are associated with the development of pulmonary arterial hypertension (PAH) include: connective tissue disorders like scleroderma, systemic lupus erythematosus (SLE), critical congenital heart disease, or Down syndrome. Researchers have also identified nongenetic factors that increase the risk of developing pulmonary arterial hypertension. These include certain drugs used as appetite suppressants and several illegal drugs, such as cocaine and methamphetamine. Pulmonary arterial hypertension is also a rare complication of certain infectious diseases such as HIV and schistosomiasis or associated with other medical conditions like cirrhosis of the liver and congenital heart diseases.
The most common cause of genetic pulmonary arterial hypertension (heritable pulmonary arterial hypertension) is mutations in the BMPR2 (bone morphogenetic protein receptor type 2) gene 59, 60, 61, 62, 63. The bone morphogenetic protein receptor type 2 (BMPR2) gene belongs to a family of genes originally identified for its role in regulating the growth and maturation (differentiation) of bone and cartilage 64. The BMPR2 gene provides instructions for making a protein called bone morphogenetic protein receptor type 2. Recently, researchers have found that the BMPR2 gene family plays a broader role in regulating the growth and differentiation of numerous types of cells. Bone morphogenetic protein receptor type 2 spans the cell membrane, so that one end of the protein is on the outer surface of the cell and the other end remains inside the cell. This positioning allows the protein to receive and transmit signals that help the cell respond to its environment by growing and dividing (cell proliferation) or by undergoing controlled cell death (apoptosis). This balance of cell proliferation and apoptosis regulates the number of cells in tissues.
Researchers suggest that a mutation in the BMPR2 gene promotes cell division or prevents cell death, resulting in an overgrowth of cells in small arteries throughout the lungs. As a result, these arteries narrow in diameter, which increases the resistance to blood flow. Blood pressure in the pulmonary artery and the right ventricle of the heart increases to overcome the increased resistance to blood flow.
Mutations in several additional genes have also been found to cause genetic pulmonary arterial hypertension (heritable pulmonary arterial hypertension), but they are much less common causes of pulmonary arterial hypertension (PAH) than are BMPR2 gene mutations. Variations in other genes may increase the risk of developing pulmonary arterial hypertension or modify the course of pulmonary arterial hypertension (PAH) usually making it more severe. Changes in as-yet-unidentified genes may also be associated with genetic pulmonary arterial hypertension.
Although pulmonary arterial hypertension often occurs on its own, it can also be part of syndromes that affect many parts of the body. For example, pulmonary arterial hypertension (PAH) is occasionally found in people with scleroderma, systemic lupus erythematosus (SLE), critical congenital heart disease, or Down syndrome.
Researchers have also identified nongenetic factors that increase the risk of developing pulmonary arterial hypertension. These include certain drugs used as appetite suppressants and several illegal drugs, such as cocaine and methamphetamine. Pulmonary arterial hypertension is also a rare complication of certain infectious diseases, including HIV and schistosomiasis.
Table 2. Pulmonary arterial hypertension genetic mutations
| Gene | Pulmonary hypertension phenotypic association | Putative molecular mechanism | Inheritance pattern | Potential distinguishing clinical and examination features | Investigations | Populations |
|---|---|---|---|---|---|---|
| BMPR2 | Heritable and idiopathic PAH | Haploinsufficiency | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Children and adult |
| ATP13A3 | Heritable and idiopathic PAH | Unknown | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Adult |
| AQP1 | Heritable and idiopathic PAH | Unknown | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Adult |
| ABCC8 | Heritable and idiopathic PAH | Haploinsufficiency | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Adult |
| KCNK3 | Heritable and idiopathic PAH | Haploinsufficiency | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Adult |
| SMAD9 | Heritable and idiopathic PAH | Haploinsufficiency | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Adult |
| Sox17 | Heritable and idiopathic PAH Congenital heart disease | Unknown | Autosomal dominant | No specific or diagnostic clinical features described | No discriminative investigations described | Children and adult |
| CAV1 | Heritable and idiopathic PAH Lipodystrophy | Gain of function; dominant negative | Autosomal dominant | Deficiency of subcutaneous adipose tissue | Fasting triglyceride and leptin levels | Children and adult |
| TBX4 | Heritable and idiopathic PAH Small patella syndrome (ischiopatellar dysplasia) Parenchymal lung disease Bronchopulmonary dysplasia Persistent pulmonary hypertension of the neonate | Unknown | Autosomal dominant | Patellar aplasia Skeletal abnormalities, in particular pelvis, knees, and feet | Skeletal X-rays: pelvis, knees, and feet CT chest: diffuse parenchymal lung disease | Children and (less commonly) adult |
| EIF2AK4 | Pulmonary veno-occlusive disease/pulmonary capillary haemangiomatosis | Loss of function | Autosomal recessive | Distal phalangeal clubbing | Reduced DLCO CT chest: interlobular septal thickening and mediastinal lymphadenopathy, and centrilobular ground-glass nodular opacities | Adult |
| KDR | Heritable and idiopathic PAH | Loss of function | Autosomal dominant | No specific or diagnostic clinical features described | Possible reduced DLCO | Older-onset adult |
| ENG | Heritable and idiopathic PAH Hereditary haemorrhagic telangiectasia | Unknown | Autosomal dominant | Telangiectasia | Iron-deficiency anaemia | Adult and children |
| ACVRL1 | Heritable and idiopathic PAH Hereditary haemorrhagic telangiectasia | Haploinsufficiency | Autosomal dominant | Abnormal blood vessel formation | Presence on imaging of pulmonary, hepatic, cerebral, or spinal arteriovenous malformations | Adult and children |
| GDF2 | Heritable and idiopathic PAH Hereditary haemorrhagic telangiectasia | Haploinsufficiency | Autosomal dominant | Visceral arteriovenous malformations Bleeding diathesis | Invasive endoscopic assessment of gastrointestinal telangiectasia | Adult and children |
Abbreviations: CT = computed tomography; DLCO = lung diffusion capacity for carbon monoxide; PAH = pulmonary arterial hypertension
[Source 58 ]Pulmonary arterial hypertension inheritance pattern
Pulmonary arterial hypertension (APH) is usually sporadic, which means it occurs in individuals with no known family history of the disorder. These non-familial cases are described as idiopathic pulmonary arterial hypertension. About 20 percent of these cases are caused by mutations in one of the genes known to be associated with pulmonary arterial hypertension, but most of the time a causative gene mutation has not been identified.
Inherited pulmonary arterial hypertension cases are known as familial pulmonary arterial hypertension. When pulmonary arterial hypertension (PAH) is inherited, it most often has an autosomal dominant pattern of inheritance, which means one copy of an altered gene in each cell is sufficient to cause pulmonary arterial hypertension. However, many people with an altered gene never develop pulmonary arterial hypertension; this phenomenon is called reduced penetrance.
Risk factors for developing pulmonary arterial hypertension
Risk factors for developing pulmonary arterial hypertension (PAH) include:
- Connective tissue disease.
- Down syndrome.
- Family history of pulmonary hypertension.
- HIV.
- Smoking.
- Use of diet medications such as “fen-phen” (dexfenfluramine and phentermine).
- Use of street drugs such as cocaine or methamphetamine.
- Blood-clotting disorders or a family history of blood clots in the lungs.
- Exposure to asbestos.
- A heart problem that you’re born with, called a congenital heart defect.
- Being overweight.
If someone in your biological family has pulmonary arterial hypertension (PAH), ask your doctor about genetic testing. If you’re diagnosed with pulmonary arterial hypertension (PAH), you may want to tell your family members so they can consider genetic testing.
Pulmonary arterial hypertension signs and symptoms
In the early stages of pulmonary arterial hypertension (PAH), you may not notice any symptoms at all. As the disease progresses, you will start to experience symptoms common to other lung diseases.
The most common symptoms of pulmonary arterial hypertension (PAH) are:
- Shortness of breath with exertion (exertional dyspnea) and eventually while at rest.
- Fatigue
- Edema, or swelling of your feet, legs and eventually your abdomen and neck
- Dizziness and fainting spells
- Chest pressure or pain.
- Fast pulse or pounding heartbeat (racing or pounding)
- Blue or gray skin color due to low oxygen levels. Your lips and fingers turning blue. But depending on your skin color, these changes may be harder or easier to see.
- Cough
- Hoarseness.
Shortness of breath is the most common symptom of pulmonary hypertension. But it may be caused by other health conditions such as asthma. Because symptoms are similar to other common lung diseases, it can often be hard to diagnose pulmonary arterial hypertension (PAH). See a doctor for an accurate diagnosis.
Pulmonary arterial hypertension complications
Pulmonary arterial hypertension potential complications include:
- Right-sided heart enlargement and heart failure. Also called cor pulmonale, this condition causes the heart’s right lower chamber to get larger. The chamber has to pump harder than usual to move blood through narrowed or blocked lung arteries. As a result, the heart walls thicken. The right lower heart chamber stretches to increase the amount of blood it can hold. These changes create more strain on the heart, and eventually the right lower heart chamber fails.
- Pleural effusions. Pleural effusion, which some people call “water on the lungs”, is the buildup of excess fluid in the pleural space, the area between the lungs and the chest wall. The pleura are thin membranes that line your lungs and the inside of your chest cavity. Normally, a small amount of fluid in the pleural space helps the lungs and chest wall to move smoothly during breathing. Pleural effusion occurs when the body produces too much fluid, doesn’t absorb enough, or when fluid leaks into the pleural space.
- Ascites. Ascites is fluid buildup in the peritoneal cavity, causing abdominal swelling. A swollen belly and gaining weight are symptoms of ascites.
- Blood clots. Having pulmonary hypertension increases the risk of blood clots in the small arteries in the lungs.
- Irregular heartbeats. Pulmonary hypertension can cause changes in the heartbeat, called arrhythmias, which can be life-threatening.
- Bleeding in the lungs. Pulmonary hypertension can lead to life-threatening bleeding into the lungs and coughing up blood.
- Pregnancy complications. Pulmonary hypertension can be life-threatening for the mother and the developing baby.
- Death. The most common cause of death in pulmonary arterial hypertension (PAH) is right ventricular failure.
Pulmonary arterial hypertension diagnosis
Pulmonary arterial hypertension is hard to diagnose early because it’s not often found during a routine physical exam. Even when pulmonary arterial hypertension is more advanced, its symptoms are similar to those of other heart and lung conditions.
To diagnose pulmonary arterial hypertension, a doctor examines you and asks about your symptoms. You’ll likely be asked questions about your medical and family history.
Tests
With the help of lung and heart specialists (pulmonologist and cardiologist) you will need to complete several tests, such as:
- Blood tests. Blood tests can help find the cause of pulmonary hypertension or show signs of complications. Brain natriuretic peptide (BNP) and N-terminal pro-brain natriuretic peptide (NT-proBNP) remain the only biomarkers routinely used in clinical practice at pulmonary hypertension centers, correlating with myocardial stress and providing prognostic information 65. Brain natriuretic peptide (BNP) and N-terminal pro-brain natriuretic peptide (NT-proBNP) are not specific for pulmonary hypertension, as they can be elevated in other forms of heart disease, exhibiting great variability. The previously proposed cut-off levels of BNP (<50, 50–300, and >300 ng/L) and NT-proBNP (<300, 300–1400, and >1400 ng/L) for low, intermediate, and high risk, respectively, in the European Society of Cardiology and European Respiratory Society risk-assessment model at baseline and during follow-up are prognostic for long-term outcomes and can be used to predict response to treatment 66. Refined cut-off values for BNP (<50, 50–199, 200–800, and >800 ng/L) and NT-pro-BNP (<300, 300–649, 650–1100, and >1100 ng/L) for low, intermediate–low, intermediate–high, and high risk, respectively, have recently been introduced as part of a four-strata risk-assessment strategy 67.
- Chest X-ray. A chest X-ray creates pictures of the heart, lungs and chest. It may be used to check for other lung conditions that can cause pulmonary hypertension. Chest radiography presents abnormal findings in most patients with pulmonary hypertension; however, a normal chest X-ray does not exclude pulmonary hypertension 68. Radiographic signs of pulmonary hypertension include a characteristic configuration of the cardiac silhouette due to right heart (right atrium [RA]/RV) and PA enlargement, sometimes with pruning of the peripheral vessels. In addition, signs of the underlying cause of pulmonary hypertension, such as left heart disease or lung disease, may be found 69, 70, 71, 72, 73.
- Electrocardiogram (ECG or EKG). This simple test records the electrical activity of the heart. It can show changes in the heartbeat.
- Typical ECG abnormalities in pulmonary hypertension:
- P pulmonale (P>0.25 mV in lead II)
- Right or sagittal axis deviation (QRS axis >90° or indeterminable)
- Right ventricle hypertrophy (R/S >1, with R >0.5 mV in V1; R in V1 + S in lead V5 >1 mV)
- Right bundle branch block—complete or incomplete (qR or rSR patterns in V1)
- Right ventricular strain patterna (ST depression/T-wave inversion in the right precordial V1–4 and inferior II, III, aVF leads)
- Prolonged QTc interval (unspecific). Patients with pulmonary arterial hypertension can present with a prolonged QTc interval (although non-specific), which may reflect right ventricular dysfunction and delayed myocardial repolarization, and is an independent predictor of mortality
- Typical ECG abnormalities in pulmonary hypertension:
- Echocardiogram. Sound waves are used to create moving images of the beating heart. An echocardiogram shows blood flow through the heart. This test may be done to help diagnose pulmonary hypertension or to determine how well treatments are working. Sometimes, an echocardiogram is done while exercising on a stationary bike or treadmill to learn how activity affects the heart. If you have this test, you may be asked to wear a mask that checks how well the heart and lungs use oxygen and carbon dioxide.
- Right heart catheterization. If an echocardiogram shows pulmonary hypertension, right heart catheterization may be done to confirm the diagnosis. This test is invasive, so it is not usually performed unless other tests cannot produce a firm diagnosis. During right heart catheterization procedure (cardiac cath or heart cath), a cardiologist places a thin, flexible tube called a catheter into a blood vessel, usually in your neck. The catheter is gently guided into the lower right heart chamber and the pulmonary artery. A doctor can then measure blood pressure in the main pulmonary arteries and the right ventricle.
- Cardiac catheterization of the right heart to measure accurate pressures (at end-expiration) is the gold standard test. It measures pulmonary artery pressure, left ventricular filling pressure, and cardiac output. Idiopathic PAH (IPAH) patients have high mean pulmonary artery pressures (greater than 25 mmHg at rest and greater than 30 mmHg with exercise) with normal pulmonary capillary wedge pressure (18 and below) 2. Pericardial effusion confers poor prognosis.
- Patients with pulmonary artery hypertension on cardiac catheterization should undergo vasoreactivity testing with short-acting pulmonary vasodilatory medications at the same time as cardiac catheterization. A decrease in the mean pulmonary artery pressure by at least 10 mmHg to reach an absolute value of 40 mmHg or less without a decrease in cardiac output. This is important as the patients who respond to these drugs can be treated with calcium channel blockers and have a more favorable prognosis 2.
- Vasoreactivity testing. The purpose of vasoreactivity testing in pulmonary arterial hypertension (PAH) is to identify acute vasoresponders who may be candidates for treatment with high-dose calcium channel blockers. Pulmonary vasoreactivity testing is only recommended in patients with idiopathic pulmonary arterial hypertension (IPAH), hereditary pulmonary arterial hypertension (HPAH), or drug- or toxin-associated pulmonary arterial hypertension. Inhaled nitric oxide or inhaled iloprost are the recommended test compounds for vasoreactivity testing 74, 75, 76. There is similar evidence for intravenous (I.V.) epoprostenol, but due to incremental dose increases and repetitive measurements, testing takes much longer and is therefore less feasible 74. Adenosine i.v. is no longer recommended due to frequent side effects 77. A positive acute response is defined as a reduction in mPAP by ≥10 mmHg to reach an absolute value ≤40 mmHg, with increased or unchanged cardiac output 74. In patients with pulmonary hypertension associated with left heart disease (Group 2), vasoreactivity testing is restricted to evaluating heart transplantation candidacy, and in patients with pulmonary hypertension in the context of congenital heart disease with initial systemic-to-pulmonary shunting, vasoreactivity testing can be performed to evaluate the possibility of defect closure 78.
Other tests may be done to check the condition of the lungs and pulmonary arteries. Doctors diagnose pulmonary hypertension based on the blood pressure in your pulmonary arteries. The criteria for diagnosis is pulmonary arterial pressure higher than 20 mmHg while you’re at rest. A right heart catheterization measures this number.
The following tests may give more information about the cause of pulmonary hypertension:
- Exercise stress tests. These tests often involve walking on a treadmill or riding a stationary bike while the heartbeat is watched. They can show how the heart reacts to exercise.
- Heart CT scan, called a cardiac CT scan. Computerized tomography (CT) scan uses X-rays to create cross-sectional images of specific parts of your heart. Dye called contrast may be given into a vein to help the blood vessels show up more clearly on the images. Cardiac CT scan can show the size of your heart and any blockages in the pulmonary arteries. It can help diagnose lung diseases that might lead to pulmonary hypertension such as chronic obstructive pulmonary disease (COPD) or pulmonary fibrosis.
- Cardiac magnetic resonance imaging (MRI) also called heart MRI scan. In a cardiac MRI, you lie on a table inside a long tube-like machine that produces a magnetic field. The magnetic field aligns atomic particles in some of your cells. When radio waves are broadcast toward these aligned particles, they produce signals that vary according to the type of tissue they are. The signals create images of your heart. It can show blood flow in the pulmonary arteries and determine how well the right lower heart chamber is working.
- Pulmonary exercise tests: Pulmonary exercise tests measure how well your lungs work when you are active. A technician will administer the test and monitor you the whole time to track your heart rate, blood pressure, oxygen and carbon dioxide levels, and your breathing patterns while active. This type of pulmonary function test is used to:
- evaluate your warning signs or symptoms like shortness of breath
- monitor your lung disease
- assess your need for supplemental oxygen
- measure your risk before a surgical procedure
- understand how your body responds to activity
- 6-Minute Walk Test: You will walk at your normal pace for six minutes. This test most often can take place in a long hallway. The 6-Minute Walk Test objectively measures how far you can walk and to see if your oxygen levels drop when you are physically active. This test will monitor your body’s response to treatments for heart, lung, and other health problems.
- Exercise tests or stress tests: Stress tests measure how your heart and blood vessels respond to exertion. You will walk on a treadmill or ride a stationary bike to perform the test while attached to an ECG machine. You will start with a warmup, then the technician will increase your incline or resistance. You will want to continue the test until you are exhausted then slowly cool down. Following the test, you will rest and continue to be monitored by the technician. Stress tests help doctors see if you have coronary artery disease. Stress tests also determine how well your body is responding to your heart’s decreased pumping effectiveness and can help guide long-term treatment decisions. If your doctor also wants to see images of your heart while you’re exercising, he or she may order a nuclear stress test or a stress echocardiogram. It’s similar to an exercise stress test, but it also uses imaging techniques to visualize your heart during the test.
- Lung function test. For this test, you blow into a special device. The device measures how much air the lungs can hold. It shows how air flows in and out of the lungs.
- Sleep study also known as polysomnography. A sleep study measures brain activity, heart rate, blood pressure, oxygen levels and other things as you sleep. The test can help diagnose sleep apnea, which can cause pulmonary hypertension.
- Ventilation/perfusion (V/Q) scan. In this test, a radioactive tracer is given through a vein (IV). The tracer shows blood flow. You also may breathe in a tracer that shows airflow to the lungs. A V/Q scan can show whether blood clots are causing symptoms of pulmonary hypertension. Pulmonary ventilation/perfusion (VQ) scan can rule out chronic thromboembolic pulmonary hypertension.
- Lung biopsy. Rarely, a sample of tissue may be taken from the lung to check for a possible cause of pulmonary hypertension.
- Genetic testing. A DNA test (genetic testing) is a medical test that can identify mutations in your genes, chromosomes or proteins. Genetic testing looks for changes in your genes, chromosomes and proteins. DNA tests can give you lots of information about the genes that make up who you are. They can confirm if you have or don’t have a specific disease. Screening for gene changes that cause pulmonary hypertension may be recommended. If you have these gene changes, other family members may need to be screened too. Genetic counselling by appropriately trained pulmonary arterial hypertension geneticists should be performed prior to genetic testing, to address the complex questions related to penetrance, genetically at-risk family members, reproduction, genetic discrimination, and psychosocial issues. Careful genetic counselling with genetic counsellors or medical geneticists is critical prior to genetic testing for asymptomatic family members 79. If the familial mutation is known and an unaffected family member tests negative for that mutation, the risk of pulmonary arterial hypertension for that person is the same as for the general population 79.
Pulmonary arterial hypertension differential diagnosis
Pulmonary arterial hypertension differential diagnosis is broad since the symptoms are non-specific:
- Chronic asthma
- Anemia
- Heart failure
- Chronic obstructive pulmonary disease (COPD)
- Cor pulmonale
- Dilated cardiomyopathy
- Mitral stenosis
- Connective tissue diseases
- Portal hypertension
- Obstructive sleep apnea (OSA)
Pulmonary hypertension functional classification
Once a diagnosis of pulmonary hypertension is confirmed, the condition is classified according to how the symptoms affect you and your ability to do everyday tasks.
World Health Organization’s (WHO) Functional Class for pulmonary hypertension may fall into one of the following groups:
- WHO Functional Class 1. Pulmonary hypertension is diagnosed, but there are no symptoms during rest or exercise. Ordinary physical activity does not cause undue shortness of breath (dyspnea) or fatigue, chest pain, or near syncope (fainting)
- WHO Functional Class 2. Patients with pulmonary hypertension with no symptoms at rest. Everyday chores or activities such as going to work or the grocery store may cause some shortness of breath (dyspnea) or fatigue, chest pain, or near syncope. There’s a slight limitation of physical activity.
- WHO Functional Class 3. Patients with pulmonary hypertension is comfortable at rest, but doing simple tasks such as bathing, dressing or preparing meals causes fatigue, shortness of breath (dyspnea) and chest pain or near syncope. The ability to do physical activity becomes very limited.
- WHO Functional Class 4. Patients with pulmonary hypertension with an inability to carry out any physical activity without symptoms. These patients manifest signs of right heart failure. Dyspnea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity.
The World Health Organization functional class is one of the strongest predictors of survival, both at diagnosis and follow-up, and worsening World Health Organization functional class is one of the most alarming indicators of disease progression, which should trigger further investigations to identify the cause(s) of clinical deterioration 80, 81, 82, 83, 84.
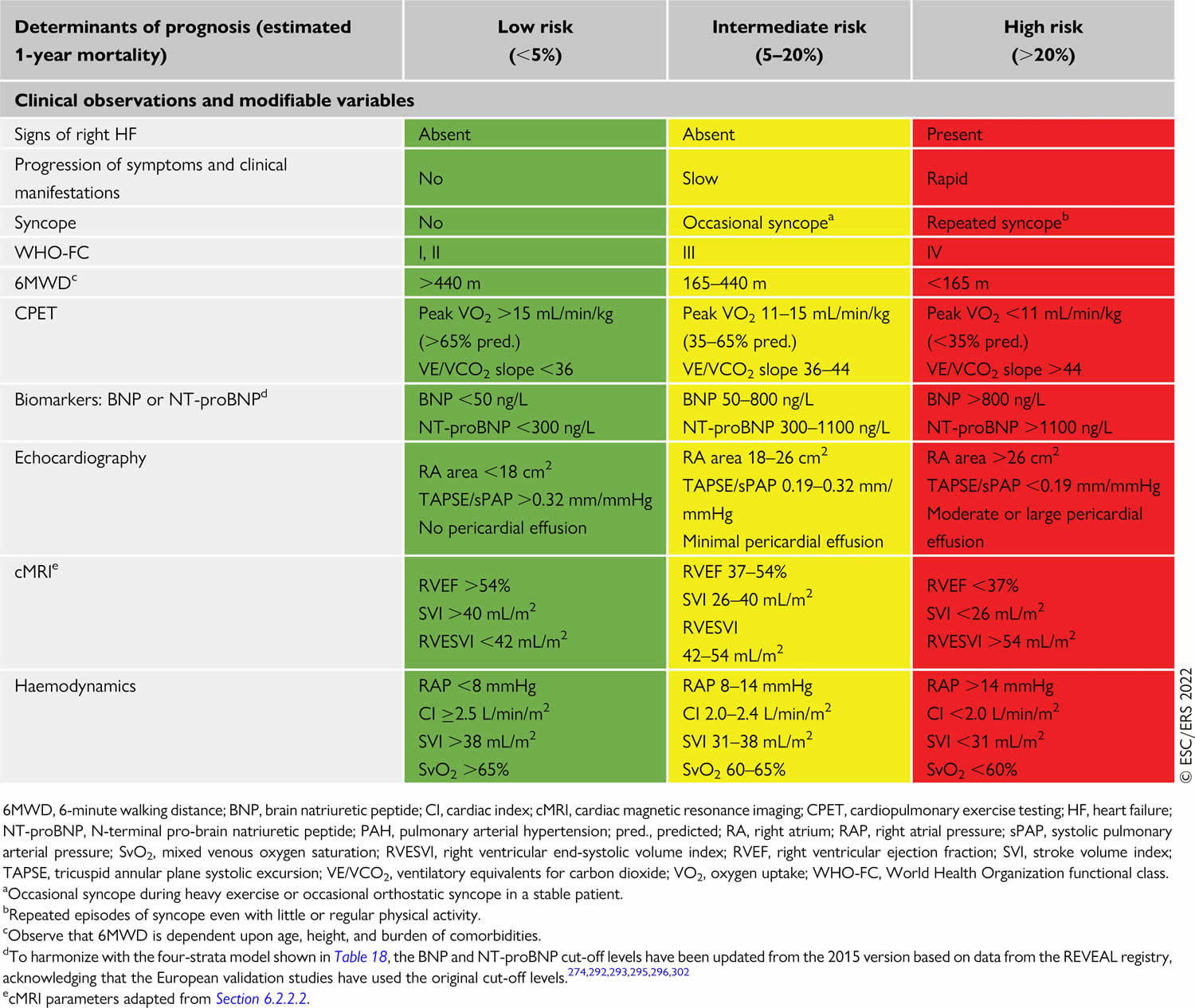
Your doctor may also use a risk calculator that considers your symptoms and test results to understand what type of treatment is needed. This is called pulmonary hypertension risk stratification.
Table 3. Pulmonary hypertension risk stratification
Pulmonary arterial hypertension treatment
Treatment options vary from person to person and depends on the cause, the type and severity of the pulmonary arterial hypertension. If you are newly diagnosed with pulmonary arterial hypertension (PAH), you should be referred to an accredited pulmonary hypertension care center for thorough evaluation. Because pulmonary arterial hypertension (PAH) is such a rare disease, it is extremely valuable to see a specialist at an accredited center to ensure you are getting the most up-to-date treatment options.
The management of pulmonary arterial hypertension (PAH) is based on New York Heart Association (NYHA) Functional Classification (i.e., patient symptoms and functional status) with the goal of positive impact on the quality of life by improving symptoms and functional status.
The New York Heart Association (NYHA) Functional Classification is a system that categorizes heart failure patients based on their symptoms and physical activity limitations, ranging from Class I (no limitation) to Class IV (symptoms at rest).
Here’s a breakdown of the NYHA classifications. New York Heart Association (NYHA) classification system groups heart failure into four categories by number. You may see Roman numerals used for these category names.
- Class 1 (I) heart failure. There are no heart failure symptoms.
- Class 2 (II) heart failure. Everyday activities can be done without difficulty. But exertion causes shortness of breath or fatigue.
- Class 3 (III) heart failure. It’s difficult to complete everyday activities.
- Class 4 (IV) heart failure. Shortness of breath occurs even at rest. This category includes the most severe heart failure.
Lifestyle and home remedies
Lifestyle changes may help improve pulmonary hypertension symptoms. Try these tips:
- Eat healthy. Eat a healthy diet rich in whole grains, fruits and vegetables, lean meats, and low-fat dairy products. Try to stay away from saturated fat, trans fat and cholesterol. Limit salt.
- Stay as active as possible and manage your weight. Even mild forms activity might be too exhausting for some people who have pulmonary hypertension. For others, moderate exercise, such as walking, might be helpful — especially when done during oxygen therapy. Your health care team can help you plan an appropriate exercise program.
- Don’t smoke. If you smoke, the most important thing you can do for your heart and lungs is to stop. If you need support quitting, ask your health care team for treatment that can help. Avoid secondhand smoke too, if possible.
- Get plenty of rest. Resting can reduce tiredness related to pulmonary hypertension.
- Avoid high altitudes. High altitudes can make pulmonary hypertension worse. If you live at an altitude of 8,000 feet (2,438 meters) or higher, you might be told to consider moving to a lower altitude.
- Avoid activities that can excessively lower blood pressure. These include sitting in a hot tub or sauna or taking long hot baths or showers. Such activities lower blood pressure and can cause fainting or even death. Also, do not do activities that cause a lot of straining, such as lifting heavy objects or weights.
- Give your health care team a list of your medicines. Some medicines can make pulmonary hypertension worse or affect its treatment.
- Get regular health checkups. Many pulmonary hypertension care centers require visits every few months and regular testing such as echocardiograms and 6-minute walk testing. Your doctor may also have you complete cardiopulmonary exercise testing (CPET), a specialized type of exercise test that measures your exercise ability. Some centers will do right heart catheterizations every year to see how well treatment is working on managing pulmonary pressures and heart function. Additionally, it is essential to take your medications exactly as directed, being careful not to run out or change your schedule unless directed by your doctor.
- Get recommended vaccines. Respiratory infections can cause serious health concerns for people with pulmonary hypertension. Ask your health care team about recommend vaccines to prevent common viral infections.
- Talk to your doctor before becoming pregnant. Pregnancy can put strain on your body and for a woman with pulmonary arterial hypertension (PAH) be possibly life-threatening, so this is a subject you should discuss with your doctor prior to becoming pregnant. Pulmonary hypertension can cause serious complications to both mother and baby during pregnancy. Birth control pills can increase the risk of blood clots. Talk to your doctor about other birth control options.
Medications
If you have pulmonary arterial hypertension (PAH), you may get medicines to treat your symptoms and help you feel better. Medicines also may be used to treat or prevent complications. Pulmonary arterial hypertension (PAH) specific medications come in multiple forms: oral, inhaled and subcutaneous (meaning delivered by an injection or IV). The medicines for pulmonary arterial hypertension (PAH) work in a few ways. Some allow blood to flow more easily through the arteries of your lungs. Others help your heart and lungs work better.
Pulmonary arterial hypertension (PAH) specific medications aim to restore balance among one or more of three substances that are produced by your lungs: nitric oxide, endothelin, and prostacyclin. Although a test does not currently exist to determine which of these substances is not balanced, pulmonary arterial hypertension (PAH) medications act on these three pathways to help slow how quickly your disease worsens.
Table 4. Pulmonary arterial hypertension medications
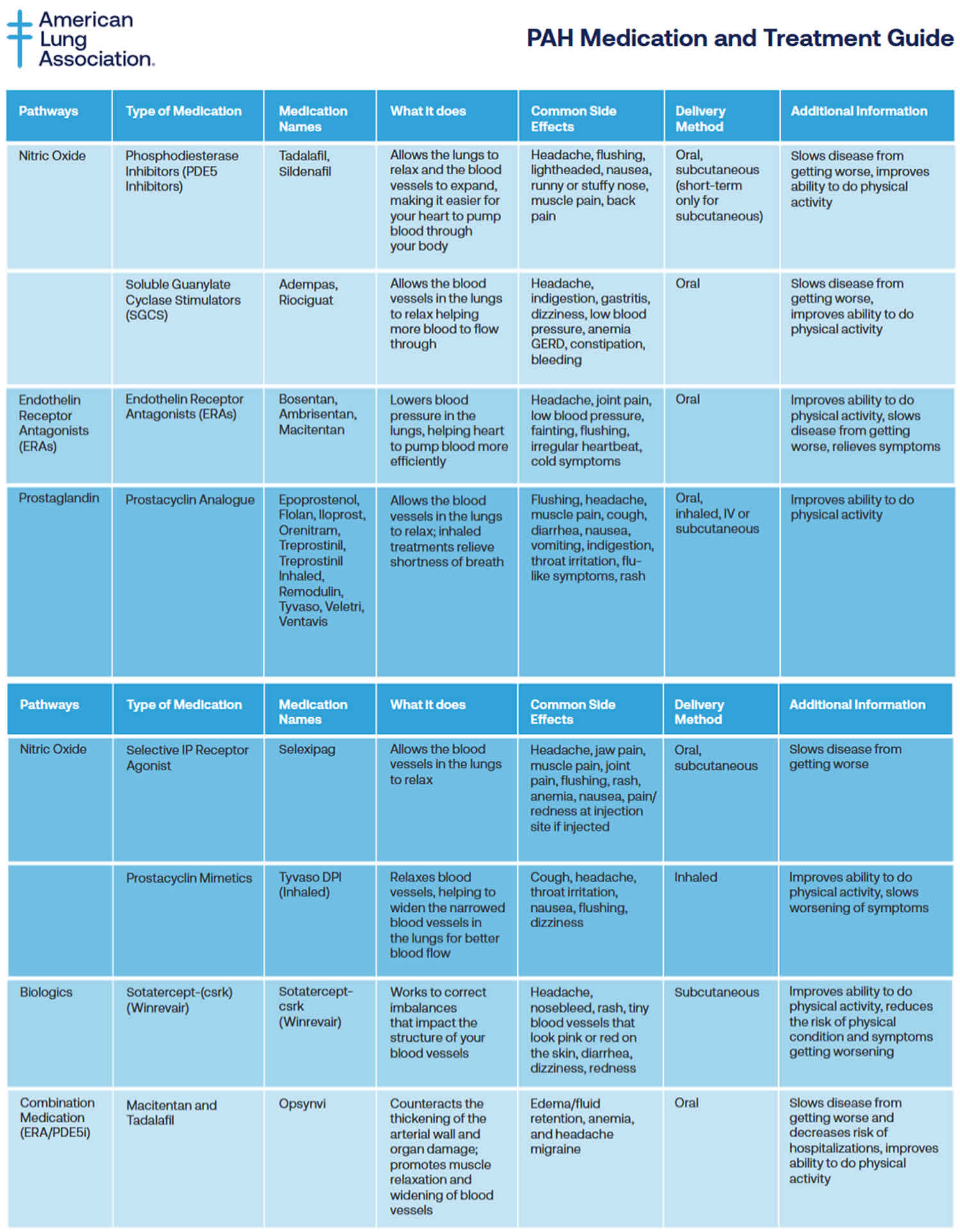
Pulmonary arterial hypertension (PAH) specific medications may include:
If you have pulmonary hypertension, you may get medicines to treat your symptoms and help you feel better. Medicines also may be used to treat or prevent complications. Treatment may include:
- Medicines to relax blood vessels (prostaglandins or prostacyclins) also called vasodilators, these medicines help open narrowed blood vessels and improve blood flow. The medicine comes in many forms. It may be breathed in, taken by mouth or given by IV. Some types are given continuously through a small pump attached to the body. Examples of vasodilators to treat pulmonary hypertension include epoprostenol (Flolan, Veletr), treprostinil (Remodulin, Tyvaso, others), Iloprost (Ventavis) and selexipag (Uptravi).
- Inhaled treatments to relieve shortness of breath and improve your ability to do physical activity. Prostacyclin analogues include Epoprostenol, Flolan, Iloprost, Orenitram, Treprostinil, Treprostinil Inhaled, Remodulin, Tyvaso, Veletri, Ventavis. Delivery Method: Oral, inhaled, IV or subcutaneous. Common Side Effects: Flushing, headache, muscle pain, cough, diarrhea, nausea, vomiting, indigestion, throat irritation, flu-like symptoms, rash.
- Selective nonprostanoid IP prostacyclin receptor agonist called Selexipag (Uptravi) targets the prostacyclin pathway by helping to prevent clotting in the arteries and by slowing the thickening of blood vessels 86. Selexipag comes as a tablet to take by mouth. It is usually taken with food twice a day. There is also injection form of Selexipag (Uptravi). Common Side Effects: Headache, jaw pain, muscle pain, joint pain, flushing, rash, anemia, nausea, pain/redness at injection site if injected.
- Tyvaso DPI (Inhaled). Tyvaso DPI is a drug-device combination therapy comprised of a small, portable, reusable, breath-powered, dry powder inhaler (DPI) for the delivery of treprostinil. Common Side Effects: Cough, headache, throat irritation, nausea, flushing, dizziness.
- Soluble guanylate cyclase (sGC) stimulators. This type of medicine relaxes the pulmonary arteries and lowers pressure in the lungs. Examples include riociguat (Adempas). Do not take these medicines if you’re pregnant.
- Medicines to widen blood vessels. Medicines called endothelin receptor antagonists (ERAs) reverse the effect of a substance in the walls of blood vessels that causes them to narrow. Such medicines include bosentan (Tracleer), macitentan (Opsumit) and ambrisentan (Letairis). They may improve energy level and symptoms. Do not take these medicines if you are pregnant.
- Medicines to increase blood flow. Medicines called phosphodiesterase 5 (PDE5) inhibitors may be used to increase blood flow through the lungs. These medicines also are used to treat erectile dysfunction. They include sildenafil (Revatio, Viagra) and tadalafil (Adcirca, Alyq, Cialis).
- Sotatercept-csrk (Winrevair) is in a class of medications called activin signaling inhibitors. Sotatercept-csrk (Winrevair) works by blocking certain substances to slow or stop the tissue changes that happen with pulmonary arterial hypertension (PAH). Sotatercept-csrk (Winrevair) can improve your ability to exercise and perform your usual activities with fewer symptoms. It can also reduce the risk of your physical condition and symptoms worsening. Sotatercept-csrk comes as a solution (liquid) to inject subcutaneously (just under the skin). It is usually given once every 3 weeks. Common Side Effects: Headache, nosebleed, rash, tiny blood vessels that look pink or red on the skin, diarrhea, dizziness, redness.
- Macitentan and Tadalafil (Opsynvi) combination works by relaxing smaller blood vessels in your lungs and increasing the supply of blood to your lungs, which reduces the workload of your heart. Macitentan and Tadalafil (Opsynvi) is only available for female patients under a restricted distribution program called the Macitentan-Containing Products REMS (Risk Evaluation and Mitigation Strategy) program. Male patients do not need to enroll in the REMS program. Macitentan is in a class of medications called endothelin receptor antagonists (ERAs). Macitentan works by stopping the action of endothelin, a natural substance that causes blood vessels to narrow and prevents normal blood flow in people who have PAH. Tadalafil is in a class of medications called phosphodiesterase (PDE) inhibitors. Tadalafil works by relaxing the blood vessels in the lungs to allow blood to flow more easily. Macitentan and tadalafil comes as a tablet to take by mouth. It is usually taken with or without food once a day. Common Side Effects: Edema/fluid retention, anemia, and headache/migraine.
- High-dose calcium channel blockers. These medicines help relax the muscles in the walls of blood vessels. They include amlodipine (Norvasc), diltiazem (Cardizem, Tiazac, others) and nifedipine (Procardia). Although calcium channel blockers can be effective, only a small number of people with pulmonary hypertension improve while taking them.
- Blood thinners also called anticoagulants, these medicines help prevent blood clots. One example is warfarin (Jantoven). Blood-thinning medicines slow the clotting process. The medicines can increase the risk of bleeding. This is especially true if you’re having surgery or a procedure that enters the body or creates an opening in the skin. Talk to your doctor about your risk.
- Digoxin (Lanoxin) also called digitalis, helps the heart squeeze better to pump blood. It also tends to slow the heartbeat. Digoxin reduces heart failure symptoms in people with heart failure with reduced ejection fraction (HFrEF). It may be more likely to be given to someone with a heart rhythm disorder, such as atrial fibrillation. The level of digoxin in the body must be checked using a blood test. If too much digoxin builds up in your blood, side effects may occur, including loss of appetite, nausea, vomiting and headaches. The heart rhythm can also become too fast or too slow. Always report any side effects of Digoxin to your doctor right away.
- Water pills also called diuretics. These medicines help the kidneys remove excess fluid from the body. This reduces the amount of work the heart has to do. Diuretics also may be used to reduce fluid buildup in the lungs, legs and belly area. Some diuretics make the body lose potassium and magnesium. Your doctor may recommend supplements to treat this. If you’re taking a diuretic, you may have regular blood tests to check your potassium and magnesium levels.
- Oxygen therapy. Breathing pure oxygen is sometimes recommended as a treatment for pulmonary hypertension. This treatment may be suggested if you live at a high altitude or have sleep apnea. Some people with pulmonary hypertension need oxygen therapy all the time.
Calcium channel blockers and the vasoactive substance are mainly used for idiopathic pulmonary hypertension (IPAH). Many new agents have been introduced, and their effectiveness can be measured by a “6-minute walk test” 87, 88, 89. Oral, high-dose calcium channel blockers (diltiazem, nifedipine) are the first-line treatment but used only in those with vasoreactivity testing positive for acute vasodilator response with short-acting pulmonary vasodilators such as adenosine, nitric oxide, or epoprostenol. The criteria for testing positive is a fall in pulmonary artery pressure to more than 10 mmHg with an increase or no change in cardiac output. Although first-line useful only in 5% of patients with idiopathic pulmonary hypertension (IPAH) and should not be used in non-responders to vasoreactivity test due to the risk of harm rather than any improvement 2. Vasoactive substances such as endothelin receptor antagonists, phosphodiesterase inhibitors, and prostanoids alter the mechanisms causing pulmonary artery smooth muscle proliferation and contraction 2.
For New York Heart Association (NYHA) Functional Class 2:
- Oral endothelin receptor antagonists (ambrisentan, bosentan, macitentan), macitentan, and modified bosentan have been shown to reduce morbidity and mortality in some studies.
- Phosphodiesterase type-5 inhibitors, PDE5 inhibitors (sildenafil, tadalafil) relax arterial smooth muscles and pulmonary artery vasodilation while inhibiting vascular remodeling.
- Non-parenteral prostanoids can be added.
For New York Heart Association (NYHA) Functional Class 3, Class 4, and those unresponsive to previous therapies:
Prostanoid agents (epoprostenol, treprostinil, iloprost): Continuous long-term intravenous epoprostenol infusion for which a semi-permanent central venous catheter is required is considered the most effective therapy. It has been shown to improve mortality, but a short half-life and high cost are the limitations. For those who cannot tolerate intravenous infusion, inhaled or subcutaneous prostanoids can be considered. Treprostinil can be used by various routes such as intravenous, subcutaneous, and inhalation. Oral prostanoids are still under clinical trials. Benefits include vasodilation, platelet inhibition, antiproliferative, and inotropic effects 90.
Soluble guanylate cyclase stimulators (riociguat, cinaciguat) are under clinical trials and are beneficial in pulmonary artery hypertension as they have a dual mode of action. They stimulate the receptor to mimic nitric oxide action and increase the sensitivity of guanylyl cyclase to endogenous nitric oxide. Riociguat has been shown to improve exercise capacity and decrease pulmonary vascular resistance in the studies.
Selexipag, a newer drug, is a selective IP prostacyclin receptor agonist. Monotherapy does relieve symptoms in patients with PAH but has not been shown to improve the prognosis and survival, and this has led to a shift to combination therapy which has shown improvement in survival (especially combination therapy including prostaglandins).
Combination therapy is being used now with the goal of targeting different mechanisms involved in the pathogenesis of PAH simultaneously (prostacyclin, endothelin, and nitric oxide pathways). A combination is considered better than increasing the dose of a single drug used and has better outcomes. Although the most common combination used is ERAs and PDE5 inhibitors, which have also been shown to reduce hospitalization in a study, but with newer drugs available now, other combinations can also be used.
Some observational studies suggest an improvement in survival in primary pulmonary hypertension with long-term anticoagulation. Also, based on the symptoms and with progression to heart failure, certain other drugs like diuretics, digoxin, and oxygen can be added.
Table 5. Dosing of pulmonary arterial hypertension medication in adults
| Starting dose | Target dose | |
|---|---|---|
| Calcium channel blockers | ||
| Amlodipine | 5 mg o.d. | 15–30 mg o.d.a |
| Diltiazem | 60 mg b.i.d.b | 120–360 mg b.i.d.b |
| Felodipine | 5 mg o.d. | 15–30 mg o.d.a |
| Nifedipine | 10 mg t.i.d. | 20–60 mg b.i.d. or t.i.d. |
| Endothelin receptor antagonists (oral administration) | ||
| Ambrisentan | 5 mg o.d. | 10 mg o.d. |
| Bosentan | 62.5 mg b.i.d. | 125 mg b.i.d. |
| Macitentan | 10 mg o.d. | 10 mg o.d. |
| Phosphodiesterase 5 inhibitors (oral administration) | ||
| Sildenafil | 20 mg t.i.d. | 20 mg t.i.d.c |
| Tadalafil | 20 or 40 mg o.d. | 40 mg o.d. |
| Prostacyclin analogues (oral administration) | ||
| Beraprost sodium | 20 µg t.i.d. | Maximum tolerated dose up to 40 µg t.i.d. |
| Beraprost extended release | 60 µg b.i.d. | Maximum tolerated dose up to 180 µg b.i.d. |
| Treprostinil | 0.25 mg b.i.d. or 0.125 mg t.i.d. | Maximum tolerated dose |
| Prostacyclin receptor agonist (oral administration) | ||
| Selexipag | 200 µg b.i.d. | Maximum tolerated dose up to 1600 µg b.i.d. |
| Soluble guanylate cyclase stimulator (oral administration) | ||
| Riociguat d | 1 mg t.i.d. | 2.5 mg t.i.d. |
| Prostacyclin analogues (inhaled administration) | ||
| Iloprost e | 2.5 µg 6–9 times per day | 5.0 µg 6–9 times per day |
| Treprostinil e | 18 µg 4 times per day | 54–72 µg 4 times per day |
| Prostacyclin analogues (i.v. or s.c. administration) | ||
| Epoprostenol i.v. | 2 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 16–30 ng/kg/min, with wide individual variability |
| Treprostinil s.c. or i.v. | 1.25 ng/kg/min | Determined by tolerability and effectiveness; typical dose range at 1 year is 25–60 ng/kg/min, with wide individual variability |
Abbreviations: b.i.d. = twice daily; i.v. = intravenous; o.d., once daily; s.c. = subcutaneous; t.i.d. = three times daily.
Footnotes: Dosages are those commonly used in clinical practice. This does not exclude the use of alternative dosages.
a The daily dosages of amlodipine and felodipine can be administered in a single dose or divided into two doses.
b There are different release formulations of diltiazem, some of which should be administered o.d. or t.i.d.
c Sildenafil is approved at a dose of 20 mg t.i.d. but doses used in practice vary widely and are sometimes higher.
d In patients at risk of systemic hypotension, riociguat may be started at 0.5 mg t.i.d.
e Doses provided are for nebulizers and may differ with the use of other formulations and other inhalation devices.
Patient follow up
The optimal timing of follow-up right heart catheterization has not been determined. While some centers regularly perform invasive follow-up assessments, others perform them as clinically indicated, and there is no evidence that any of these strategies is associated with better outcomes.
Table 6. Pulmonary arterial hypertension follow-up
Pulmonary rehabilitation
Pulmonary rehabilitation is a supervised six – eight week exercise and education program that helps people with chronic lung diseases improve their breathing and overall well-being through exercise, education, and behavioral changes. Pulmonary rehabilitation may improve your exercise endurance, muscle strength and quality of life.
Pulmonary rehabilitation is an outpatient program and may be based in a hospital or a clinic. You may also be able to receive certain forms of pulmonary rehabilitation in your own home.
To find a pulmonary rehabilitation program in your area, visit:
Surgery and other procedures
If medicines do not help control the symptoms of pulmonary hypertension, surgery may be recommended 91. Surgeries and procedures to treat pulmonary hypertension may include:
- Atrial septostomy. This treatment may be recommended if medicines don’t control pulmonary hypertension symptoms. In an atrial septostomy, a doctor creates an opening between the upper left and right chambers of your heart. The opening reduces the pressure on the right side of the heart. Potential complications include irregular heartbeats called arrhythmias.
- Lung transplant or heart-lung transplant. Sometimes, a lung or heart-lung transplant may be needed, especially for younger people who have idiopathic pulmonary arterial hypertension. After a transplant, medicine must be taken for life to help reduce the chance of rejection.
Pulmonary arterial hypertension prognosis
Studies indicate that early diagnosis of pulmonary hypertension is associated with improved survival rates. Your life expectancy depends on many factors, including the severity of your pulmonary hypertension and how early you’re diagnosed. Talk with your doctor to learn your specific prognosis. Untreated patients have very poor survival, but some can survive into the 5th decade of life with medical treatment 92, 93. Patients who fail to respond to medications usually have the worst prognosis. Patients with persistently elevated pulmonary pressures and right heart failure usually are dead within 5 years 94, 89.
The Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL registry) PAH Risk Score Calculator is a tool clinicians may use to help prognosticate patients with pulmonary artery hypertension. The tool has validation as a predictive algorithm for 1-year survival.
You can use the PAH Risk Score Calculator here: https://pahriskcalculatorre.com/
Factors that are independently associated with decreased survival include the following:
- Men older than 60 years
- PAH associated with portal hypertension or connective tissue disorder
- Family history of PAH
- WHO Class 3 or 4 kidney insufficiency
- Resting systolic blood pressure less than 110 mmHg
- Heart rate greater than 92 beats per minute
- Six-minute walk test less than 165 m
- Brain natriuretic peptide (BNP) greater than 180 pg/ml
- Pulmonary vascular resistance greater than 32 Woods units
- Presence of pericardial effusion on echocardiogram
- TAPSE (tricuspid annular plane systolic excursion) less than 1.5 cm, percentage predicted diffusing capacity of the lung for carbon monoxide (DLCO less than 32%)
The prognosis of idiopathic pulmonary hypertension (IPAH) is poor 2. The mean survival of untreated idiopathic pulmonary hypertension (IPAH) is 2 to 3 years from the diagnosis. The New York Heart Association (NYHA) functional class is an important predictor of survival, with class 4 mean survival of less than 6 months 2. The most important prognostic factor is right ventricular function which is also the cause of death in advanced idiopathic pulmonary hypertension (IPAH). Increased mortality is seen in pregnant patients with advanced idiopathic pulmonary hypertension (IPAH).
In another study, patients with scleroderma with PAH compared with patients with idiopathic pulmonary hypertension (IPAH) have a threefold higher mortality risk 95, 96, 97, 98. Furthermore, patients scleroderma with PAH stand out from those with other types of PAH, given their impaired response to traditional therapies and worse overall clinical prognosis despite exhibiting similar end-organ pathology and often presenting with milder hemodynamic impairment 95, 99. Proposed factors explaining these disparities include more pronounced inflammation, autoimmunity, the distinct nature of the underlying vasculopathy, and differing abilities of the right ventricle to adapt to the increased afterload 100, 101, 102.
Steps you can take to improve your prognosis (outlook) include:
- Create an emergency kit. You need to have certain supplies and information with you all the time. Ask your doctor what you should include in your kit, and never leave home without it.
- Get your vaccines as your doctor recommends. It’s important to protect yourself against the flu and pneumonia.
- Keep all your follow-up appointments. Regular testing is crucial to check your lung and heart function and measure treatment progress.
- Take your medications as your doctor prescribes, and at the same time every day. Don’t make any changes to your medication routine unless your doctor tells you to do so.
- Krowl L, Anjum F, Kaul P. Pulmonary Idiopathic Hypertension. [Updated 2023 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK519041[↩][↩]
- Pahal P, Sharma S. Idiopathic Pulmonary Artery Hypertension. [Updated 2023 Apr 10]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482251[↩][↩][↩][↩][↩][↩][↩][↩]
- Oldroyd SH, Manek G, Bhardwaj A. Pulmonary Hypertension. [Updated 2024 May 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482463[↩][↩]
- Humbert M, Kovacs G, Hoeper MM, Badagliacca R, et al. ESC/ERS Scientific Document Group. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J. 2022 Oct 11;43(38):3618-3731. doi: 10.1093/eurheartj/ehac237. Erratum in: Eur Heart J. 2023 Apr 17;44(15):1312. doi: 10.1093/eurheartj/ehad005[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Gaine S, Chin K, Coghlan G, Channick R, Di Scala L, Galie N, et al. Selexipag for the treatment of connective tissue disease-associated pulmonary arterial hypertension. Eur Respir J. 2017;50:1602493. doi: 10.1183/13993003.02493-2016[↩]
- Kato M, Atsumi T. Pulmonary arterial hypertension associated with connective tissue diseases: a review focusing on distinctive clinical aspects. Eur J Clin Invest. 2018 doi: 10.1111/eci.12876[↩]
- Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, et al. Pulmonary arterial hypertension in France: results from a national registry. Am J Resp Crit Care. 2006;173:1023–1030. doi: 10.1164/rccm.200510-1668OC[↩]
- McLaughlin VV, Hoeper MM, Channick RN, Chin KM, Delcroix M, Gaine S, Ghofrani HA, Jansa P, Lang IM, Mehta S, Pulido T, Sastry BKS, Simonneau G, Sitbon O, Souza R, Torbicki A, Tapson VF, Perchenet L, Preiss R, Verweij P, Rubin LJ, Galiè N. Pulmonary Arterial Hypertension-Related Morbidity Is Prognostic for Mortality. J Am Coll Cardiol. 2018 Feb 20;71(7):752-763. doi: 10.1016/j.jacc.2017.12.010[↩]
- Chapter 57 – Pulmonary Hypertension in Non-Pulmonary Arterial Hypertension Patients. Vascular Medicine: A Companion to Braunwald’s Heart Disease (Second Edition) 2013, Pages 687-696. https://doi.org/10.1016/B978-1-4377-2930-6.00057-4[↩]
- Galiè N, McLaughlin VV, Rubin LJ, Simonneau G. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019 Jan 24;53(1):1802148. doi: 10.1183/13993003.02148-2018[↩]
- Vachiéry JL, Tedford RJ, Rosenkranz S, Palazzini M, Lang I, Guazzi M, Coghlan G, Chazova I, De Marco T. Pulmonary hypertension due to left heart disease. Eur Respir J. 2019 Jan 24;53(1):1801897. doi: 10.1183/13993003.01897-2018[↩]
- Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, Williams PG, Souza R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019 Jan 24;53(1):1801913. doi: 10.1183/13993003.01913-2018[↩][↩]
- Widrich J, Shetty M. Physiology, Pulmonary Vascular Resistance. [Updated 2024 Jan 31]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK554380[↩]
- Pulmonary arterial hypertension. https://medlineplus.gov/genetics/condition/pulmonary-arterial-hypertension[↩]
- Learn About Pulmonary Arterial Hypertension. https://www.lung.org/lung-health-diseases/lung-disease-lookup/pulmonary-arterial-hypertension/learn-about-pulmonary-arterial-hypertension[↩][↩]
- Badesch D.B., Raskob G.E., Elliott C.G., et al. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376–387. doi: 10.1378/chest.09-1140[↩]
- Virsinskaite R, Karia N, Kotecha T, Schreiber BE, Coghlan JG, Knight DS. Pulmonary hypertension – the latest updates for physicians. Clin Med (Lond). 2023 Sep;23(5):449-454. doi: 10.7861/clinmed.2023-23.5.Cardio4[↩][↩]
- Zanatta E, Polito P, Famoso G, Larosa M, De Zorzi E, Scarpieri E, Cozzi F, Doria A. Pulmonary arterial hypertension in connective tissue disorders: Pathophysiology and treatment. Exp Biol Med (Maywood). 2019 Feb;244(2):120-131. doi: 10.1177/1535370218824101[↩]
- Berteloot L, Proisy M, Jais JP, Lévy M, Boddaert N, Bonnet D, Raimondi F. Idiopathic, heritable and veno-occlusive pulmonary arterial hypertension in childhood: computed tomography angiography features in the initial assessment of the disease. Pediatr Radiol. 2019 May;49(5):575-585. doi: 10.1007/s00247-018-04331-y[↩]
- Weiss BM, Zemp L, Seifert B, Hess OM. Outcome of pulmonary vascular disease in pregnancy: a systematic overview from 1978 through 1996. J Am Coll Cardiol. 1998 Jun;31(7):1650-7. doi: 10.1016/s0735-1097(98)00162-4[↩]
- Bédard E, Dimopoulos K, Gatzoulis MA. Has there been any progress made on pregnancy outcomes among women with pulmonary arterial hypertension? Eur Heart J. 2009 Feb;30(3):256-65. doi: 10.1093/eurheartj/ehn597[↩]
- Duarte AG, Thomas S, Safdar Z, Torres F, Pacheco LD, Feldman J, deBoisblanc B. Management of pulmonary arterial hypertension during pregnancy: a retrospective, multicenter experience. Chest. 2013 May;143(5):1330-1336. doi: 10.1378/chest.12-0528[↩]
- Jaïs X, Olsson KM, Barbera JA, Blanco I, Torbicki A, Peacock A, Vizza CD, Macdonald P, Humbert M, Hoeper MM. Pregnancy outcomes in pulmonary arterial hypertension in the modern management era. Eur Respir J. 2012 Oct;40(4):881-5. doi: 10.1183/09031936.00141211[↩][↩]
- Kiely DG, Condliffe R, Webster V, Mills GH, Wrench I, Gandhi SV, Selby K, Armstrong IJ, Martin L, Howarth ES, Bu’lock FA, Stewart P, Elliot CA. Improved survival in pregnancy and pulmonary hypertension using a multiprofessional approach. BJOG. 2010 Apr;117(5):565-74. doi: 10.1111/j.1471-0528.2009.02492.x[↩][↩]
- Luo J, Shi H, Xu L, Su W, Li J. Pregnancy outcomes in patients with pulmonary arterial hypertension: A retrospective study. Medicine (Baltimore). 2020 Jun 5;99(23):e20285. doi: 10.1097/MD.0000000000020285[↩]
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015 Oct;46(4):903-75. doi: 10.1183/13993003.01032-2015 Erratum in: Eur Respir J. 2015 Dec;46(6):1855-6. doi: 10.1183/13993003.51032-2015[↩]
- Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016 Jan 1;37(1):67-119. doi: 10.1093/eurheartj/ehv317[↩]
- Kamp JC, von Kaisenberg C, Greve S, Winter L, Park DH, Fuge J, Kühn C, Hoeper MM, Olsson KM. Pregnancy in pulmonary arterial hypertension: Midterm outcomes of mothers and offspring. J Heart Lung Transplant. 2021 Mar;40(3):229-233. doi: 10.1016/j.healun.2020.12.002[↩][↩]
- Corbach N, Berlier C, Lichtblau M, Schwarz EI, Gautschi F, Groth A, Schüpbach R, Krähenmann F, Saxer S, Ulrich S. Favorable Pregnancy Outcomes in Women With Well-Controlled Pulmonary Arterial Hypertension. Front Med (Lausanne). 2021 Jul 5;8:689764. doi: 10.3389/fmed.2021.689764[↩]
- Bostock S, Sheares K, Cannon J, Taboada D, Pepke-Zaba J, Toshner M. The potential effects of pregnancy in a patient with idiopathic pulmonary arterial hypertension responding to calcium channel blockade. Eur Respir J. 2017 Dec 14;50(6):1701141. doi: 10.1183/13993003.01141-2017[↩]
- de Raaf MA, Beekhuijzen M, Guignabert C, Vonk Noordegraaf A, Bogaard HJ. Endothelin-1 receptor antagonists in fetal development and pulmonary arterial hypertension. Reprod Toxicol. 2015 Aug 15;56:45-51. doi: 10.1016/j.reprotox.2015.06.048[↩]
- Dunn L, Greer R, Flenady V, Kumar S. Sildenafil in Pregnancy: A Systematic Review of Maternal Tolerance and Obstetric and Perinatal Outcomes. Fetal Diagn Ther. 2017;41(2):81-88. doi: 10.1159/000453062[↩]
- van Giersbergen PL, Halabi A, Dingemanse J. Pharmacokinetic interaction between bosentan and the oral contraceptives norethisterone and ethinyl estradiol. Int J Clin Pharmacol Ther. 2006 Mar;44(3):113-8. doi: 10.5414/cpp44113[↩]
- American Heart Association. About Arrhythmia. http://www.heart.org/HEARTORG/Conditions/Arrhythmia/AboutArrhythmia/About-Arrhythmia_UCM_002010_Article.jsp[↩]
- Centers for Disease Control and Prevention. Division of Birth Defects and Developmental Disabilities. Congenital Heart Defects (CHDs). https://www.cdc.gov/ncbddd/heartdefects/index.html[↩]
- National Institutes of Health. National Heart, Lung and Blood Institute. Your Heart’s Electrical System. https://www.nhlbi.nih.gov/health/health-topics/topics/hhw/electrical[↩]
- Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med 2016;4:306–22. 10.1016/S2213-2600(15)00543-3[↩][↩][↩]
- Ross H, Feingold ML, Train JS. Mesenteric venous thrombosis. N Engl J Med. 2002 Apr 18;346(16):1252-3; author reply 1252-3.[↩]
- Rosenkranz S, Gibbs JSR, Wachter R, et al. Left ventricular heart failure and pulmonary hypertension. Eur Heart J 2016;37:942–54. 10.1093/eurheartj/ehv512[↩]
- Kessler R, Faller M, Weitzenblum E, et al. “Natural history” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med 2001;164:219–24. 10.1164/ajrccm.164.2.2006129[↩]
- Blanco I, Tura-Ceide O, Peinado VI, et al. Updated perspectives on pulmonary hypertension in COPD. Int J Chron Obstruct Pulmon Dis 2020;15:1315–24. 10.2147/COPD.S211841[↩]
- Lettieri CJ, Nathan SD, Barnett SD, et al. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006;129:746–52. 10.1378/chest.129.3.746[↩]
- Nathan SD, Shlobin OA, Ahmad S, et al. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008;76:288–94. 10.1159/000114246[↩]
- Shorr AF, Wainright JL, Cors CS, et al. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J 2007;30:715–21. 10.1183/09031936.00107206[↩]
- Leber L, Beaudet A, Muller A. Epidemiology of pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: identification of the most accurate estimates from a systematic literature review. Pulm Circ 2021;11:2045894020977300. 10.1177/2045894020977300[↩]
- Delcroix M, Torbicki A, Gopalan D, et al. ERS statement on chronic thromboembolic pulmonary hypertension. Eur Respir J 2021;57:2002828. 10.1183/13993003.02828-2020[↩]
- Kramm T, Wilkens H, Fuge J, et al. Incidence and characteristics of chronic thromboembolic pulmonary hypertension in Germany. Clin Res Cardiol 2018;107:548–53. 10.1007/s00392-018-1215-5[↩]
- Shlobin OA, Kouranos V, Barnett SD, et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: results from an international registry. Eur Respir J 2020;55:1901747. 10.1183/13993003.01747-2019[↩]
- Bousseau S, Sobrano Fais R, Gu S, Frump A, Lahm T. Pathophysiology and new advances in pulmonary hypertension. BMJ Med. 2023 Mar 23;2(1):e000137. doi: 10.1136/bmjmed-2022-000137[↩]
- Zeder K, Banfi C, Steinrisser-Allex G, Maron BA, Humbert M, Lewis GD, Berghold A, Olschewski H, Kovacs G. Diagnostic, prognostic and differential-diagnostic relevance of pulmonary haemodynamic parameters during exercise: a systematic review. Eur Respir J. 2022 Oct 13;60(4):2103181. doi: 10.1183/13993003.03181-2021[↩][↩][↩]
- Ho JE, Zern EK, Lau ES, et al. Exercise pulmonary hypertension predicts clinical outcomes in patients with dyspnea on effort. J Am Coll Cardiol 2020; 75: 17–26. doi:10.1016/j.jacc.2019.10.048[↩]
- Stamm A, Saxer S, Lichtblau M, et al. Exercise pulmonary haemodynamics predict outcome in patients with systemic sclerosis. Eur Respir J 2016; 48: 1658–1667. doi:10.1183/13993003.00990-2016[↩]
- Hasler ED, Muller-Mottet S, Furian M, et al. Pressure-flow during exercise catheterization predicts survival in pulmonary hypertension. Chest 2016; 150: 57–67. doi:10.1016/j.chest.2016.02.634[↩]
- Lewis GD, Murphy RM, Shah RV, et al. Pulmonary vascular response patterns during exercise in left ventricular systolic dysfunction predict exercise capacity and outcomes. Circ Heart Fail 2011; 4: 276–285. doi:10.1161/CIRCHEARTFAILURE.110.959437[↩]
- Zeder K, Avian A, Bachmaier G, et al. Exercise pulmonary resistances predict long-term survival in systemic sclerosis. Chest 2021; 159: 781–790. doi:10.1016/j.chest.2020.08.2110[↩]
- Eisman AS, Shah RV, Dhakal BP, et al. Pulmonary capillary wedge pressure patterns during exercise predict exercise capacity and incident heart failure. Circ Heart Fail 2018; 11: e004750. doi:10.1161/CIRCHEARTFAILURE.117.004750[↩]
- Bentley RF, Barker M, Esfandiari S, et al. Normal and abnormal relationships of pulmonary artery to wedge pressure during exercise. J Am Heart Assoc 2020; 9: e016339. doi:10.1161/JAHA.120.016339[↩]
- 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. European Respiratory Journal 2023 61(1): 2200879; DOI: https://doi.org/10.1183/13993003.00879-2022[↩][↩]
- Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014 Jun 20;115(1):189-202. doi: 10.1161/CIRCRESAHA.115.303404[↩]
- Austin ED, Phillips JA III, Loyd JE. Heritable Pulmonary Arterial Hypertension Overview. 2002 Jul 18 [Updated 2020 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1485[↩]
- Machado RD, Southgate L, Eichstaedt CA, et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum Mutat. 2015 Dec;36(12):1113-27. doi: 10.1002/humu.22904[↩]
- Montani D, Günther S, Dorfmüller P, Perros F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud-Macari E, Price LC, Humbert M, Simonneau G, Sitbon O. Pulmonary arterial hypertension. Orphanet J Rare Dis. 2013 Jul 6;8:97. doi: 10.1186/1750-1172-8-97[↩]
- Soubrier F, Chung WK, Machado R, Grünig E, Aldred M, Geraci M, Loyd JE, Elliott CG, Trembath RC, Newman JH, Humbert M. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013 Dec 24;62(25 Suppl):D13-21. doi: 10.1016/j.jacc.2013.10.035[↩]
- BMPR2 gene. https://medlineplus.gov/genetics/gene/bmpr2[↩]
- Warwick G, Thomas PS, Yates DH. Biomarkers in pulmonary hypertension. Eur Respir J. 2008 Aug;32(2):503-12. doi: 10.1183/09031936.00160307[↩]
- Chin KM, Rubin LJ, Channick R, Di Scala L, Gaine S, Galiè N, Ghofrani HA, Hoeper MM, Lang IM, McLaughlin VV, Preiss R, Simonneau G, Sitbon O, Tapson VF. Association of N-Terminal Pro Brain Natriuretic Peptide and Long-Term Outcome in Patients With Pulmonary Arterial Hypertension. Circulation. 2019 May 21;139(21):2440-2450. doi: 10.1161/CIRCULATIONAHA.118.039360[↩]
- Hoeper MM, Pausch C, Olsson KM, et al. COMPERA 2.0: a refined four-stratum risk assessment model for pulmonary arterial hypertension. Eur Respir J. 2022 Jul 7;60(1):2102311. doi: 10.1183/13993003.02311-2021[↩]
- Remy-Jardin M, Ryerson CJ, Schiebler ML, et al. Imaging of pulmonary hypertension in adults: a position paper from the Fleischner Society. Eur Respir J 2021; 57: 2004455. doi:10.1183/13993003.04455-2020[↩]
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J 2015; 46: 903–975. doi:10.1183/13993003.01032-2015[↩]
- Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016; 37: 67–119. doi:10.1093/eurheartj/ehv317[↩]
- Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987; 107: 216–223. doi:10.7326/0003-4819-107-2-216[↩]
- Ascha M, Renapurkar RD, Tonelli AR. A review of imaging modalities in pulmonary hypertension. Ann Thorac Med 2017; 12: 61–73. doi:10.4103/1817-1737.203742[↩]
- Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62: D42–D50. doi:10.1016/j.jacc.2013.10.032[↩]
- Sitbon O, Humbert M, Jais X, et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 2005; 111: 3105–3111. doi:10.1161/CIRCULATIONAHA.104.488486[↩][↩][↩]
- Hoeper MM, Olschewski H, Ghofrani HA, et al. A comparison of the acute hemodynamic effects of inhaled nitric oxide and aerosolized iloprost in primary pulmonary hypertension. German PPH study group. J Am Coll Cardiol 2000; 35: 176–182. doi:10.1016/S0735-1097(99)00494-5[↩]
- Opitz CF, Wensel R, Bettmann M, et al. Assessment of the vasodilator response in primary pulmonary hypertension. Comparing prostacyclin and iloprost administered by either infusion or inhalation. Eur Heart J 2003; 24: 356–365. doi:10.1016/S0195-668X(02)00302-0[↩]
- Jing ZC, Jiang X, Han ZY, et al. Iloprost for pulmonary vasodilator testing in idiopathic pulmonary arterial hypertension. Eur Respir J 2009; 33: 1354–1360. doi:10.1183/09031936.00169608[↩]
- Baumgartner H, De Backer J, Babu-Narayan SV, et al. 2020 ESC Guidelines for the management of adult congenital heart disease. Eur Heart J 2021; 42: 563–645. doi:10.1093/eurheartj/ehaa554[↩]
- Morrell NW, Aldred MA, Chung WK, et al. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 2019; 53: 1801899. doi:10.1183/13993003.01899-2018[↩][↩]
- Barst RJ, Chung L, Zamanian RT, Turner M, McGoon MD. Functional class improvement and 3-year survival outcomes in patients with pulmonary arterial hypertension in the REVEAL Registry. Chest. 2013 Jul;144(1):160-168. doi: 10.1378/chest.12-2417[↩]
- Nickel N, Golpon H, Greer M, Knudsen L, Olsson K, Westerkamp V, Welte T, Hoeper MM. The prognostic impact of follow-up assessments in patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2012 Mar;39(3):589-96. doi: 10.1183/09031936.00092311[↩]
- Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, Rainisio M, Simonneau G. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol. 2002 Aug 21;40(4):780-8. doi: 10.1016/s0735-1097(02)02012-0[↩]
- Benza RL, Miller DP, Gomberg-Maitland M, Frantz RP, Foreman AJ, Coffey CS, Frost A, Barst RJ, Badesch DB, Elliott CG, Liou TG, McGoon MD. Predicting survival in pulmonary arterial hypertension: insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation. 2010 Jul 13;122(2):164-72. doi: 10.1161/CIRCULATIONAHA.109.898122[↩]
- Humbert M, Sitbon O, Yaïci A, Montani D, O’Callaghan DS, Jaïs X, Parent F, Savale L, Natali D, Günther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G; French Pulmonary Arterial Hypertension Network. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010 Sep;36(3):549-55. doi: 10.1183/09031936.00057010[↩]
- PAH Medication & Treatment Guide. https://www.lung.org/getmedia/db7df4fe-93fa-439b-bd24-80856b9e3365/PAH-Medication-Guide.pdf[↩]
- https://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/UPTRAVI-pi.pdf[↩]
- Behr J, Nathan SD, Harari S, Wuyts W, Kirchgaessler KU, Bengus M, Gilberg F, Wells AU. Sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: A Phase IIb, randomised, double-blind, placebo-controlled study – Rationale and study design. Respir Med. 2018 May;138:13-20. doi: 10.1016/j.rmed.2018.03.019[↩]
- Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, Kociol RD, Lewis EF, Mehra MR, Pagani FD, Raval AN, Ward C; American Heart Association Council on Clinical Cardiology; Council on Cardiovascular Disease in the Young; and Council on Cardiovascular Surgery and Anesthesia. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2018 May 15;137(20):e578-e622. doi: 10.1161/CIR.0000000000000560[↩]
- Kim D, Lee KM, Freiman MR, Powell WR, Klings ES, Rinne ST, Miller DR, Rose AJ, Wiener RS. Phosphodiesterase-5 Inhibitor Therapy for Pulmonary Hypertension in the United States. Actual versus Recommended Use. Ann Am Thorac Soc. 2018 Jun;15(6):693-701. doi: 10.1513/AnnalsATS.201710-762OC[↩][↩]
- Mishra A, Singh M, Kaluski E. The year since the guidelines: a concise update on recent advances in pulmonary hypertension. Minerva Cardioangiol. 2017 Feb;65(1):68-73. doi: 10.23736/S0026-4725.16.04240-7[↩]
- López-Meseguer M, Quezada CA, Ramon MA, Lázaro M, Dos L, Lara A, López R, Blanco I, Escribano P, Roman A; REHAP Investigators. Lung and heart-lung transplantation in pulmonary arterial hypertension. PLoS One. 2017 Nov 21;12(11):e0187811. doi: 10.1371/journal.pone.0187811. Erratum in: PLoS One. 2018 Jan 29;13(1):e0192100. doi: 10.1371/journal.pone.0192100[↩]
- Zamanian RT, Kudelko KT, Sung YK, Perez VJ, Liu J, Spiekerkoetter E. Current clinical management of pulmonary arterial hypertension. Circ Res. 2014 Jun 20;115(1):131-147. doi: 10.1161/CIRCRESAHA.115.303827[↩]
- Guillevin L, Armstrong I, Aldrighetti R, Howard LS, Ryftenius H, Fischer A, Lombardi S, Studer S, Ferrari P. Understanding the impact of pulmonary arterial hypertension on patients’ and carers’ lives. Eur Respir Rev. 2013 Dec;22(130):535-42. doi: 10.1183/09059180.00005713[↩]
- Keen C, Fowler-Davis S, McLean S, Manson J. Physiotherapy practice in pulmonary hypertension: physiotherapist and patient perspectives. Pulm Circ. 2018 Jul-Sep;8(3):2045894018783738. doi: 10.1177/2045894018783738[↩]
- Fisher M.R., Mathai S.C., Champion H.C., et al. Clinical differences between idiopathic and scleroderma-related pulmonary hypertension. Arthritis Rheum. 2006;54(9):3043–3050. doi: 10.1002/art.22069[↩][↩]
- Chung L., Liu J., Parsons L., et al. Characterization of connective tissue disease-associated pulmonary arterial hypertension from REVEAL: identifying systemic sclerosis as a unique phenotype. Chest. 2010;138(6):1383–1394. doi: 10.1378/chest.10-0260[↩]
- Ahmed S., Palevsky H.I. Pulmonary arterial hypertension related to connective tissue disease: a review. Rheum Dis Clin North Am. 2014;40(1):103–124. doi: 10.1016/j.rdc.2013.10.001[↩]
- Gashouta M.A., Humbert M., Hassoun P.M. Update in systemic sclerosis-associated pulmonary arterial hypertension. Presse Med. 2014;43(10 Pt 2):e293–e304. doi: 10.1016/j.lpm.2014.06.007[↩]
- Clements P.J., Tan M., McLaughlin V.V., et al. Investigators PAHQERIP-Q The pulmonary arterial hypertension quality enhancement research initiative: comparison of patients with idiopathic PAH to patients with systemic sclerosis-associated PAH. Ann Rheum Dis. 2012;71(2):249–252. doi: 10.1136/annrheumdis-2011-200265[↩]
- Chaisson N.F., Hassoun P.M. Systemic sclerosis-associated pulmonary arterial hypertension. Chest. 2013;144(4):1346–1356. doi: 10.1378/chest.12-2396[↩]
- Overbeek M.J., Vonk M.C., Boonstra A., et al. Pulmonary arterial hypertension in limited cutaneous systemic sclerosis: a distinctive vasculopathy. Eur Respir J. 2009;34(2):371–379. doi: 10.1183/09031936.00106008[↩]
- Kelemen B.W., Mathai S.C., Tedford R.J., et al. Right ventricular remodeling in idiopathic and scleroderma-associated pulmonary arterial hypertension: two distinct phenotypes. Pulm Circ. 2015;5(2):327–334. doi: 10.1086/680356[↩]
{kind=link}