Loeys-Dietz syndrome
Loeys-Dietz syndrome (LDS) is a inherited connective tissue disorder that causes aortic aneurysms, widely spaced eyes (hypertelorism), a uvula (the little piece of flesh that hangs down in the back of the mouth) that splits in two (bifid uvula) and/or cleft palate and tortuous arteries (twisting or spiraled arteries) 1, 2, 3. Connective tissue is a catchall term for tissues composed of collagen and elastin that hold your body together and link its different parts. Connective tissue provides strength and flexibility to structures such as tendons, ligaments, skin, cartilage, bones, fascia and blood vessels, which provide support and structure to other tissues and organs. Connective tissue is essential to maintain the structure of your body. You have different forms of connective tissue nearly everywhere in your body. Other findings in people with Loeys-Dietz syndrome (LDS) include craniosynostosis (a condition in which one or more of the fibrous sutures in a young infant’s skull prematurely fuses or closes by turning into bone too early before the brain is fully formed thereby changing the growth pattern of the skull), exotropia (eyes that turn outward), micrognathia or mandibular hypoplasia (a condition in which the lower jaw or the mandible is smaller than usual), structural brain abnormalities, intellectual deficit, and congenital heart disease 1, 2, 3. Individuals with Loeys-Dietz syndrome are also predisposed to widespread and aggressive arterial aneurysms and pregnancy-related complications including uterine rupture and death 1. Individuals with Loeys-Dietz syndrome can show a strong predisposition for allergic/inflammatory disease including asthma, eczema, and reactions to food or environmental allergens 1. People with Loeys-Dietz syndrome (LDS) also have an increased incidence of gastrointestinal inflammation including eosinophilic esophagitis and eosinophilic gastritis and inflammatory bowel disease 1.
Loeys-Dietz syndrome (LDS) was was first observed and described by Dr. Bart Loeys and Dr. Hal Dietz at the Johns Hopkins University School of Medicine in 2005 and has many shared characteristics with Marfan syndrome 2. The prevalence of Loeys-Dietz syndrome is unknown but is estimated to be 1 per 50,000 newborns 3. More than 1,000 families with Loeys-Dietz syndrome have been described in the literature until 2018. Though with more availability of genetic testing the number of patients diagnosed has increased significantly in recent years. Loeys-Dietz syndrome occurs in all ethnic groups. Loeys-Dietz syndrome is a rare disorder that affects males and females in equal numbers.
Loeys-Dietz syndrome signs and symptoms vary among individuals. There are 7 types of Loeys-Dietz syndrome, labelled types 1 through 7, which are distinguished by their genetic cause. Loeys-Dietz syndrome is a genetic disorder that is caused by a mutation (gene change) in either the TGFBR1 or TGFBR2 genes (transforming growth factor beta receptor 1 or 2), the SMAD2 or SMAD3 genes (mothers against decapentaplegic homolog 2 or 3), the TGFB2 gene (tgfbeta 2 or transforming growth factor beta 2 ligand), the TGFB3 gene (tgfbeta 3 or transforming growth factor beta 3 ligand) or the IPO8 (Importin 8) gene 1.
- Loeys-Dietz syndrome 1 caused by mutations in the TGFBR1 gene
- Loeys-Dietz syndrome 2 caused by mutations in the TGFBR2 gene
- Loeys-Dietz syndrome 3 caused by mutations in the SMAD3 gene
- Loeys-Dietz syndrome 4 caused by mutations in the TGFB2 gene
- Loeys-Dietz syndrome 5 caused by mutations in the TGFB3 gene
- Loeys-Dietz syndrome 6 caused by mutations in the SMAD2 gene
- Loeys-Dietz syndrome 7 caused by mutations in the IPO8 gene
Although there is significant overlap between the clinical features caused by mutations in the different genes, scientists are learning about what might be differing features between the types and how this may impact medical management. Loeys-Dietz syndrome types 1 and 2 appear to be the most common forms. This condition is called Loeys-Dietz syndrome type 1 when affected individuals have cleft palate, craniosynostosis, and/or hypertelorism. Individuals without these features are said to have Loeys-Dietz syndrome type 2. Regardless of the type, signs and symptoms of Loeys-Dietz syndrome can become apparent anytime from childhood through adulthood, and the severity is variable.
These seven genes, TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, TGFB3 and bi-allelic variants in IPO8, play roles in a cell signaling pathway called the transforming growth factor beta (TGF-β) pathway, which directs the functions of the body’s cells during growth and development 4. This pathway also regulates the formation of the extracellular matrix, an intricate lattice of proteins and other molecules that forms in the spaces between cells and is important for tissue strength and repair. Mutations in the TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2 or TGFB3 gene result in the production of a protein with reduced function. Even though the protein is less active, signaling within the transforming growth factor beta (TGF-β) pathway occurs at an even greater intensity than normal in tissues throughout the body. Researchers speculate that the activity of other proteins in this signaling pathway is increased to compensate for the protein whose function is reduced; however, the exact mechanism responsible for the increase in signaling is unclear. The overactive transforming growth factor beta (TGF-β) pathway disrupts the development of the extracellular matrix and various body systems, leading to the signs and symptoms of Loeys-Dietz syndrome.
In 25 percent of Loeys-Dietz syndrome cases, Loeys-Dietz syndrome is inherited in an autosomal dominant manner with variable clinical expression and an affected person inherits the mutation from one affected parent 5. However, in about 75 percent of cases, Loeys-Dietz syndrome results from a new gene mutation and occurs in people with no history of the disorder in their family. This is called a de novo mutation.
It is important to have an early and adequate treatment for the heart problems because the chance for aortic dissection and other vascular problems may be high in some patients. Aortic dissection has been observed in early childhood (age ≥6 months) and/or at aortic dimensions that do not confer risk in other connective tissue disorders such as Marfan syndrome 5. Many specialists may be involved for the best management of the patient.
In Loeys-Dietz syndrome the aorta can weaken and stretch, causing a bulge in the blood vessel wall (an aneurysm). Stretching of the aorta may also lead to a sudden tearing of the layers in the aorta wall (aortic dissection). People with Loeys-Dietz syndrome can also have aneurysms or dissections in arteries throughout the body and have arteries with abnormal twists and turns (arterial tortuosity).
Individuals with Loeys-Dietz syndrome often have skeletal problems including premature fusion of the skull bones (craniosynostosis), an abnormal side-to-side curvature of the spine (scoliosis), either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum), an inward- and upward-turning foot (clubfoot), flat feet (pes planus), or elongated limbs with joint deformities called contractures that restrict the movement of certain joints. A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia). In individuals with Loeys-Dietz syndrome, dural ectasia typically does not cause health problems. Malformation or instability of the spinal bones (vertebrae) in the neck is a common feature of Loeys-Dietz syndrome and can lead to injuries to the spinal cord. Some affected individuals have joint inflammation (osteoarthritis) that commonly affects the knees and the joints of the hands, wrists, and spine.
People with Loeys-Dietz syndrome may bruise easily and develop abnormal scars after wound healing. The skin is frequently described as translucent, often with stretch marks (striae) and visible underlying veins. Some individuals with Loeys-Dietz syndrome develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax) or a protrusion of organs through gaps in muscles (hernias). Other characteristic features include widely spaced eyes (hypertelorism), eyes that do not point in the same direction (strabismus), a split in the soft flap of tissue that hangs from the back of the mouth (bifid uvula), and an opening in the roof of the mouth (cleft palate).
Individuals with Loeys-Dietz syndrome frequently develop immune system-related problems such as food allergies, asthma, or inflammatory disorders such as eczema or inflammatory bowel disease.
There is still no cure for Loeys-Dietz syndrome. Treatment focuses on managing the specific symptoms you have. These treatments typically require a team of specialists including a geneticist, cardiologist, heart (cardiothoracic) and bone (orthopedic) surgeons, ophthalmologist, rheumatologist, among others. It is recommended that you consult your physician or a local geneticist if you have questions about your health concerns. It is always important to consult your doctor to determine an effective and personalized course of action.
Most importantly, the goal of Loeys-Dietz syndrome treatment is to screen and correct blood vessel weaknesses before they tear. Your doctor may prescribe a beta-blocker or angiotensin receptor blocker (ARB) such as losartan to slow down the ballooning or stretching of the aortic root. Every 6 months to 1 year, you should be screened for aneurysms of the entire arterial tree from your head to your hips. This screening includes blood vessel imaging by CT angiography (CTA) or MR angiography (MRA) and heart imaging by echocardiography. If repeated imaging of the entire arterial tree does not show any changes or concern for aneurysms, it may be acceptable to image less frequently. Your doctor will be looking to see whether the aortic root balloons to larger than 4 cm (1.5 inches), at which point he/she may recommend surgery to replace the ballooning section of the aorta and sometimes the aortic valve of the heart. This surgery is typically safe and effective in fixing the problem.
To treat some of the musculoskeletal abnormalities associated with Loeys-Dietz syndrome, your care team may recommend nonsurgical management of the symptoms such as bracing or surgical correction of the abnormalities. These abnormalities include an abnormally curved spine (scoliosis), an indented or protruding chest, and issues with the bones of the neck. Doctors will recommend X-rays and CT scans to determine whether surgery should be performed. These surgeries can be complicated and require close attention for complications afterwards.
Staying active and conditioned is recommended for individuals with Loeys-Dietz syndrome. Athletic goals and limits should be discussed with the cardiologist and the care team to determine an individualized health plan.
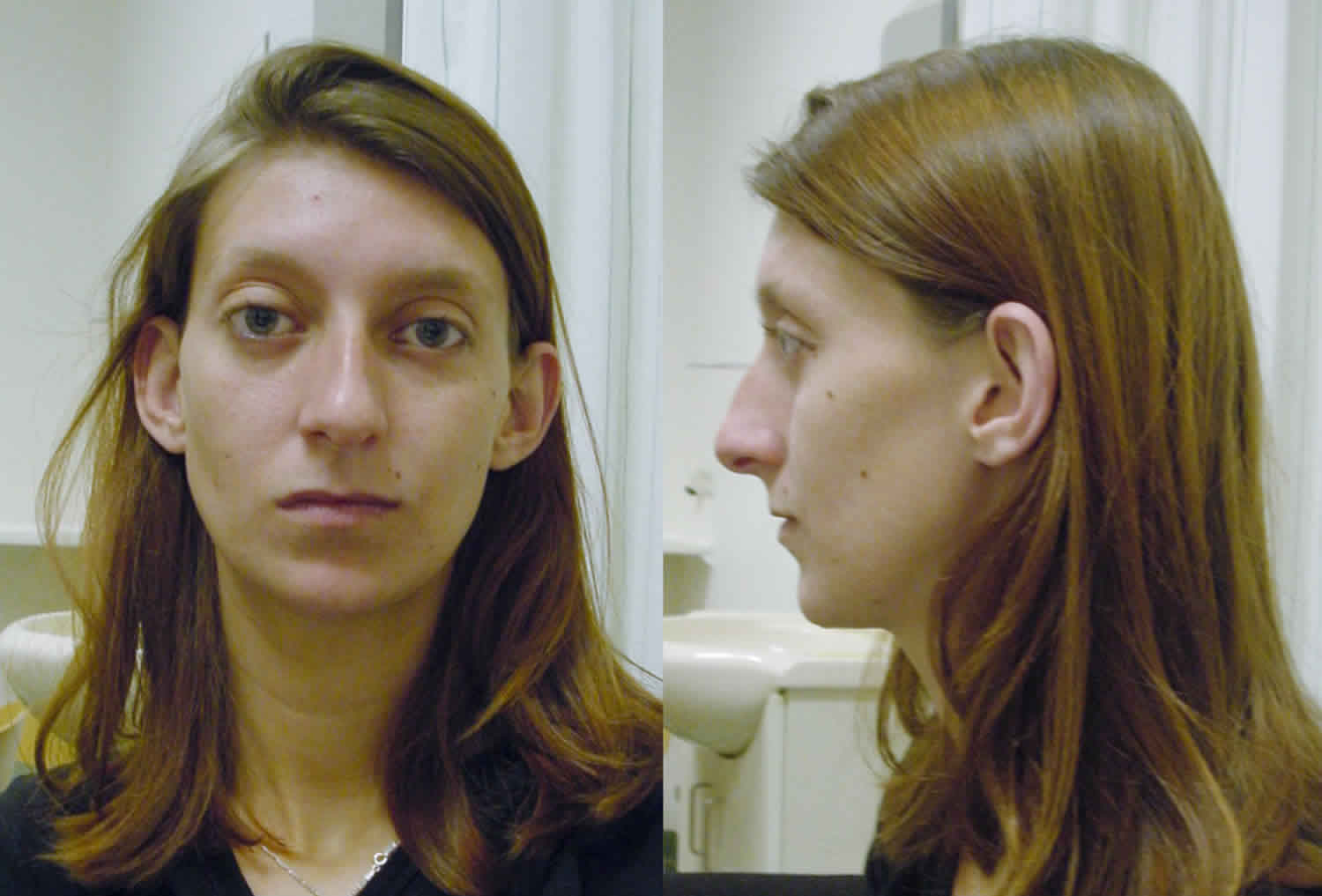
Figure 1. Loeys-Dietz syndrome features
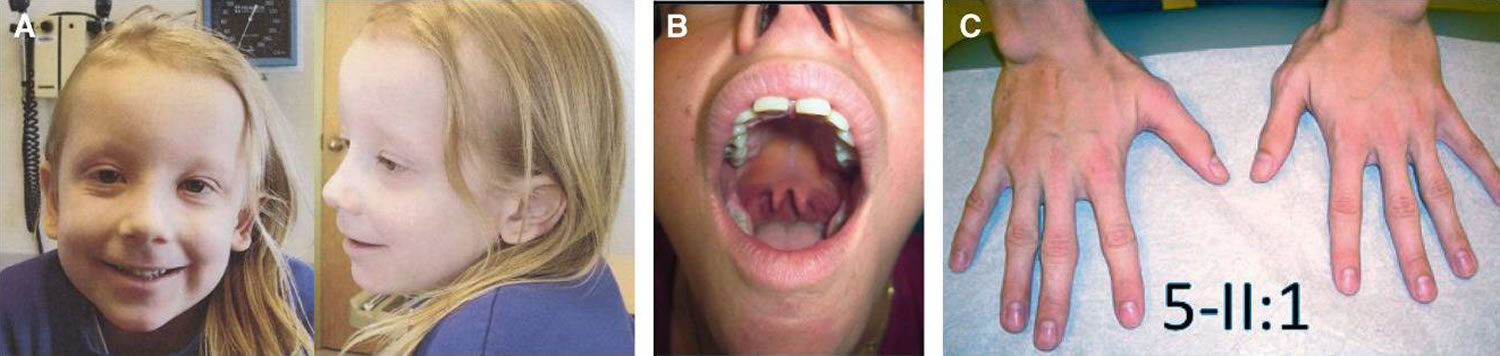
Footnotes: (A) hypertelorism, down-slanting palpebral fissures, amblyopia, and translucent skin in a 5-year-old girl, (B) bifid uvula in a 31-year-old woman, and (C) arachnodactyly in a 24-year-old man.
[Source 6 ]Figure 2. Loeys-Dietz syndrome neonatal presentation
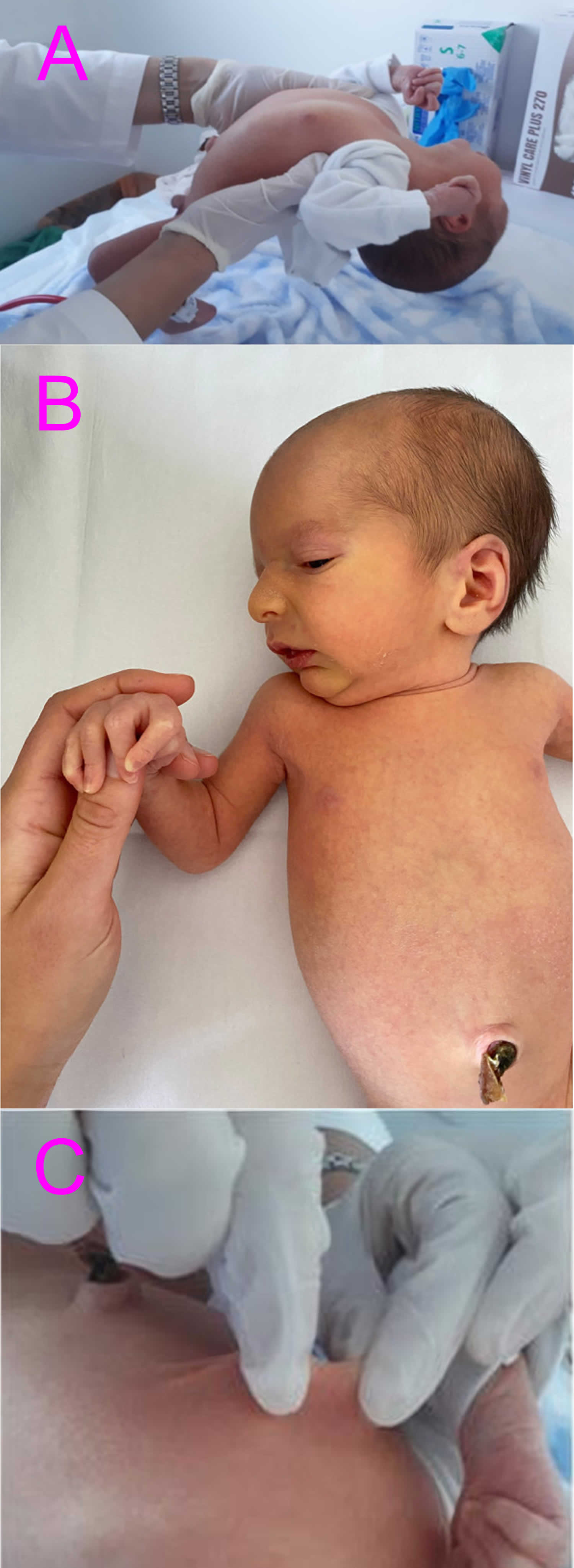
Footnotes: Loeys-Dietz syndrome neonatal presentations. (A) axial hypotonia; (B) senile appearance; (C) lax skin
[Source 7 ]Table 1. Loeys-Dietz Syndrome Associated Genes and Subtypes
| Gene | Subtype of Loeys-Dietz syndrome | Comment | Reference |
|---|---|---|---|
| TGFBR1 | Loeys-Dietz syndrome 1 | Most severe Loeys-Dietz syndrome. Loeys-Dietz syndrome caused by TGFBR1 gene or TGFBR2 gene has similar severity. | 8, 9 |
| TGFBR2 | Loeys-Dietz syndrome 2 | ||
| SMAD2 | Loeys-Dietz syndrome 6 | Variable cardiovascular Loeys-Dietz syndrome, including congenital heart disease | 10, 11 |
| SMAD3 | Loeys-Dietz syndrome 3 | Severity of aortic disease in SMAD3-related Loeys-Dietz syndrome is similar to TGFBR1- or TGFBR2-related Loeys-Dietz syndrome; strong predisposition for osteoarthritis. However, several individuals with SMAD3-related Loeys-Dietz syndrome do not have osteoarthritis 12. | 13 |
| TGFB2 | Loeys-Dietz syndrome 4 | Systemic findings possibly less severe & more like Marfan syndrome | 14, 15, 16 |
| TGFB3 | Loeys-Dietz syndrome 5 | Mildest form of Loeys-Dietz syndrome | 14, 15, 16 |
| IPO8 | Loeys-Dietz syndrome 7 | Very severe aneurysms at young age; no dissections described | 17 |
Loeys-Dietz syndrome type 1
Loeys-Dietz syndrome 1 caused by mutations in the TGFBR1 gene
Loeys-Dietz syndrome type 2
Loeys-Dietz syndrome 2 caused by mutations in the TGFBR2 gene
Loeys-Dietz syndrome type 3
Loeys-Dietz syndrome 3 caused by mutations in the SMAD3 gene
Loeys-Dietz syndrome type 4
Loeys-Dietz syndrome 4 caused by mutations in the TGFB2 gene
Loeys-Dietz syndrome type 5
Loeys-Dietz syndrome 5 caused by mutations in the TGFB3 gene
Loeys-Dietz syndrome type 6
Loeys-Dietz syndrome 6 caused by mutations in the SMAD2 gene
Loeys-Dietz syndrome type 7
Loeys-Dietz syndrome 7 caused by mutations in the IPO8 gene. IPO8-related Loeys-Dietz syndrome is inherited in an autosomal recessive manner.
Loeys-Dietz syndrome causes
The 7 types of Loeys-Dietz syndrome are distinguished by their genetic cause: TGFBR1 (transforming growth factor beta receptor 1) gene mutations cause Loeys-Dietz syndrome type 1, TGFBR2 (transforming growth factor beta receptor 2) gene mutations cause Loeys-Dietz syndrome type 2, SMAD3 (mothers against decapentaplegic homolog 3) gene mutations cause Loeys-Dietz syndrome type 3, TGFB2 (transforming growth factor beta 2 ligand) gene mutations cause Loeys-Dietz syndrome type 4, TGFB3 (transforming growth factor beta 3 ligand) gene mutations cause Loeys-Dietz syndrome type 5 and SMAD2 (mothers against decapentaplegic homolog 2) gene mutations cause Loeys-Dietz syndrome type 6 and IPO8 (importin 8) gene mutations cause Loeys-Dietz syndrome type 7 1. These 7 genes play roles in a cell signaling pathway called the transforming growth factor beta (TGF-β) pathway, which directs the functions of the body’s cells during growth and development. This pathway also regulates the formation of the extracellular matrix, an intricate lattice of proteins and other molecules that forms in the spaces between cells and is important for tissue strength and repair.
Mutations in the TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, TGFB3 or IPO8 gene result in the production of a protein with reduced function. Even though the protein is less active, signaling within the TGF-β pathway occurs at an even greater intensity than normal in tissues throughout the body. Researchers speculate that the activity of other proteins in this signaling pathway is increased to compensate for the protein whose function is reduced; however, the exact mechanism responsible for the increase in signaling is unclear. The overactive transforming growth factor beta (TGF-β) pathway disrupts the development of the extracellular matrix and various body systems, leading to the signs and symptoms of Loeys-Dietz syndrome.
Loeys-Dietz syndrome inheritance pattern
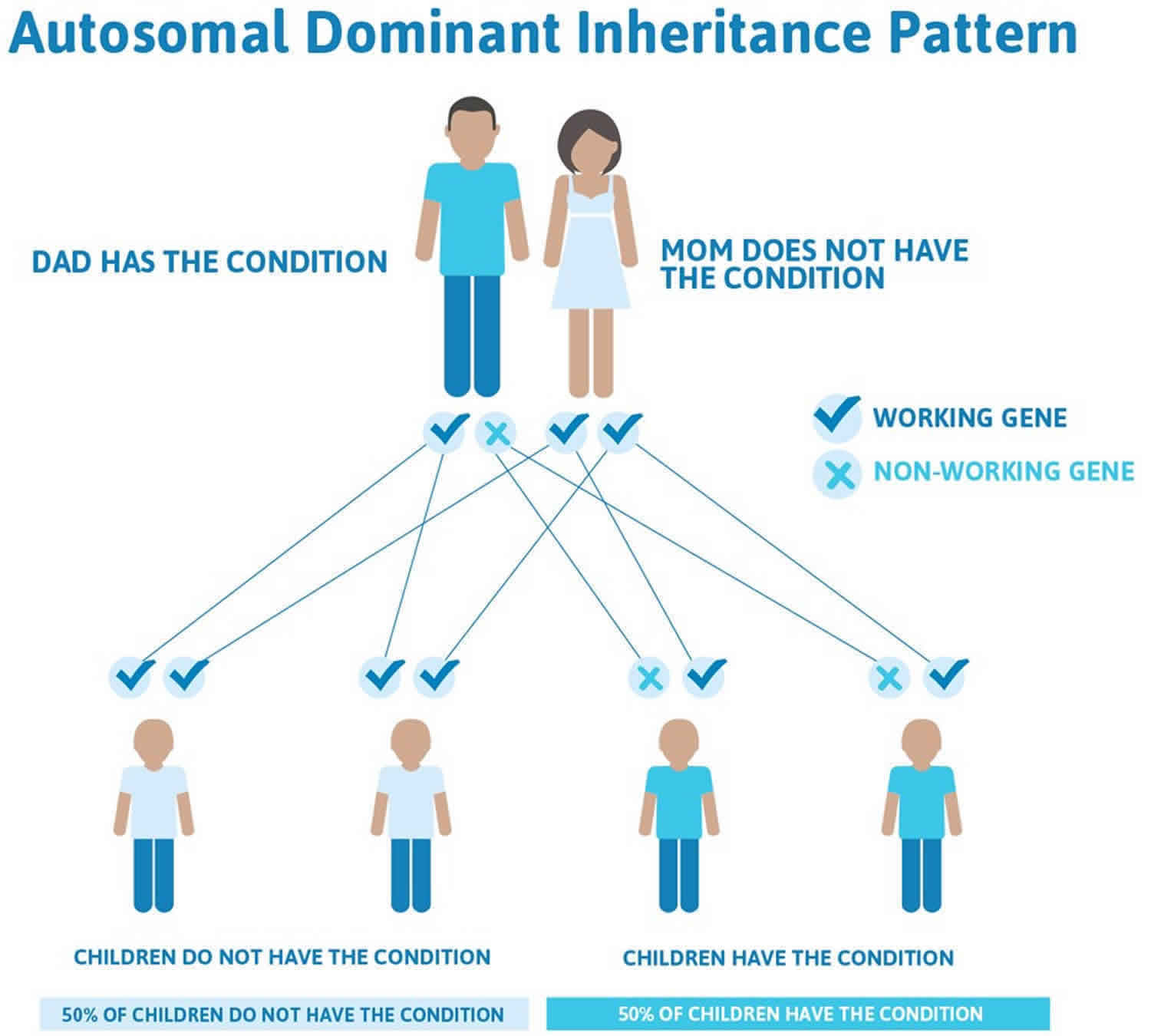
Loeys-Dietz syndrome has an autosomal dominant pattern of inheritance, which means one copy of the altered gene (TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, or TGFB3 gene) in each cell is sufficient to cause the disorder.
In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
In about 75 percent of cases, Loeys-Dietz syndrome results from a new gene mutation and occurs in people with no history of the disorder in their family. This is called a de novo mutation. In 25 percent of cases, an affected person inherits the mutation from one affected parent.
IPO8-related Loeys-Dietz syndrome type 7 is inherited in an autosomal recessive manner. An autosomal recessive inheritance is a pattern of genetic inheritance where a child needs to inherit two copies of a mutated IPO8 gene (one from each parent) which are located on the non-sex chromosomes (autosomes) to develop Loeys-Dietz syndrome type 7. The parents of a child with IPO8-related Loeys-Dietz syndrome type 7 typically do not show symptoms themselves but are “carriers” of the IPO8 gene or heterozygous for an IPO8 gene. If both parents are known to be heterozygous for an IPO8 gene, each sibling of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives requires prior identification of the IPO8 gene in the family. Males and females are affected equally.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 3. Loeys-Dietz syndrome autosomal dominant inheritance pattern
Loeys-Dietz syndrome signs and symptoms
Loeys-Dietz syndrome (LDS) is a connective tissue disorder that affects your bones, ligaments, arterial walls and skin. The symptoms can vary widely, and a person with Loeys-Dietz syndrome may only have a few of the following signs or symptoms but still have the condition. Loeys-Dietz syndrome should be suspected in individuals with the following vascular, skeletal, craniofacial, cutaneous, allergic/inflammatory, and eye findings 18. There are 4 main characteristics that suggest the diagnosis of Loeys-Dietz syndrome. These features are not usually seen all together in other connective tissue disorders as major characteristics.
4 main characteristics that suggest the diagnosis of Loeys-Dietz syndrome:
- Aneurysms (widening or dilation of arteries), which can be observed by imaging techniques. These are most often observed in the aortic root (base of the artery leading from the heart) but can be seen in other arteries throughout the body
- Aortic aneurysms (enlargement of the aorta, the main blood vessel leaving the heart)
- Aneurysms of the head/neck arteries, where the aorta exits the heart and in the abdomen, down to the popliteal area behind the knee
- About two-thirds of people with Loeys-Dietz syndrome have an aortic aneurysm at the time of diagnosis, and nearly everyone with Loeys-Dietz syndrome will have some level of aortic ballooning
- Arterial tortuosity (twisted or spiraled arteries), most often occurring in the vessels of the neck and observed on imaging techniques
- Arteries may become twisted and tortuous, increasing the risk of them splitting or dissecting
- These weakened arteries are at high risk of rupturing, which is a life-threatening emergency
- Widely spaced eyes (hypertelorism) that are more noticeable when the baby is inside the womb and in newborns or very young children
- Bifid (split) or broad uvula (the little piece of flesh that hangs down in the back of the mouth) or cleft palate (split or incomplete closure of the roof of the mouth) that may result from incomplete closing of the hard palate
It is important to note, however, that these findings are not observed in all patients and do not concretely lead to a diagnosis of Loeys-Dietz syndrome.
People with Loeys-Dietz syndrome are at risk for serious complications like ruptured aneurysms, so early detection and management are critical.
Loeys-Dietz syndrome other common signs and symptoms of may include:
- Scoliosis (abnormal sideways curve of the spine)
- Long, thin fingers resembling a spider’s legs (arachnodactyly)
- Protruded chest (pectus carinatum) or caved-in chest (pectus excavatum)
- Flat feet (pes planus)
- Club foot (a deformed foot which is twisted so that the sole cannot be placed flat on the ground)
- Joint hypermobility (very flexible joints) or joints that are too tight (in some people)
- Thin, translucent skin (veins may be easily visible through the skin)
- Early fusion of the skull (craniosynostosis) (premature closing of the skull bones) which may result in an abnormal shape of the skull and in an increased pressure inside the skull.
Loeys-Dietz syndrome is characterized by enlargement of the aorta (aortic aneurysm), which is the large blood vessel that distributes blood from your heart to the rest of your body. The aorta can weaken and stretch, causing a bulge in the blood vessel wall (an aneurysm). Stretching of the aorta may also lead to a sudden tearing of the layers in the aorta wall (aortic dissection). People with Loeys-Dietz syndrome can also have aneurysms or dissections in arteries throughout the body and have arteries with abnormal twists and turns (arterial tortuosity).
Individuals with Loeys-Dietz syndrome often have skeletal problems including premature fusion of the skull bones (craniosynostosis), an abnormal side-to-side curvature of the spine (scoliosis), either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum), an inward- and upward-turning foot (clubfoot), flat feet (pes planus), or elongated limbs with joint deformities called contractures that restrict the movement of certain joints. A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia). In individuals with Loeys-Dietz syndrome, dural ectasia typically does not cause health problems. Malformation or instability of the spinal bones (vertebrae) in the neck is a common feature of Loeys-Dietz syndrome and can lead to injuries to the spinal cord. Some affected individuals have joint inflammation (osteoarthritis) that commonly affects the knees and the joints of the hands, wrists, and spine.
People with Loeys-Dietz syndrome may bruise easily and develop abnormal scars after wound healing. The skin is frequently described as translucent, often with stretch marks (striae) and visible underlying veins. Some individuals with Loeys-Dietz syndrome develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax) or a protrusion of organs through gaps in muscles (hernias). Other characteristic features include widely spaced eyes (hypertelorism), crossed eyes or eyes that do not point in the same direction (strabismus), a split in the soft flap of tissue that hangs from the back of the mouth (bifid uvula), and an opening in the roof of the mouth (cleft palate).
Individuals with Loeys-Dietz syndrome frequently develop immune system-related problems such as food allergies, asthma, or inflammatory disorders such as eczema or inflammatory bowel disease.
Vascular
- Dilatation or dissection of the aorta and other arteries. Aortic root dilatation is seen in more than 95% of probands; the aortic root is the most common site for a dissection to occur. In rare circumstances, aneurysms or dissections can be seen in other arteries in the head, chest, abdomen, or extremities in the absence of aortic involvement.
- Other arterial aneurysms and tortuosity
- Evaluation is best done with magnetic resonance angiography (MRA) or CT angiogram (CTA) with 3D reconstruction from head to pelvis to identify arterial aneurysms or dissections and arterial tortuosity throughout the arterial tree.
- Tortuosity is often most prominent in head and neck vessels.
- Approximately 50% of individuals with Loeys-Dietz syndrome studied had an aneurysm distant from the aortic root that would not have been detected by echocardiography.
Craniofacial (head and face)
- Malar hypoplasia (flat cheek bones)
- Slight downward slant to the eyes
- Craniosynostosis (early fusion of the skull bones)
- Cleft palate (hole in the roof of the mouth)
- Blue sclerae (blue tinge to the whites of the eyes)
- Micrognathia (small chin) and/or retrognathia (receding chin)
Skeletal (bones)
- Long fingers and toes
- Contractures of the fingers
- Clubfoot or skewfoot deformity
- Scoliosis (abnormal sideways curvature of the spine)
- Cervical-spine instability (instability in the vertebrae directly below the skull)
- Joint laxity
- Pectus excavatum (chest wall deformity that causes the sternum and breast bone to grow inward) / Pectus carinatum (chest wall deformity that pushes the sternum and breast bone out)
- Osteoarthritis
- Typically normal stature
Skin
- Translucent skin
- Soft or velvety skin
- Easy bruising
- Abnormal or wide scarring
- Soft skin texture
- Hernias
- Milia, prominently on the face
Heart
- Congenital (existing at birth) heart defects, which can include patent ductus arteriosus (PDA), atrial septal defect (ASD) or ventricular septal defect (VSD) and bicuspid aortic valve.
Eyes
- Myopia (nearsightedness)
- Eye muscle disorders causing crossed eyes or strabismus
- Blue or dusky sclerae
- Retinal detachment: The retina is the light-sensitive layer of tissue that lines the inside of the eye and sends visual messages through the optic nerve to the brain. When the retina detaches, it is lifted or pulled from its normal position. If not promptly treated, retinal detachment can cause permanent vision loss.
Other signs and symptoms
- Food allergy, seasonal or environmental allergies
- Asthma or chronic sinusitis
- Eczema (dermatitis)
- Eosinophilic esophagitis/gastritis
- Inflammatory bowel disease such as Crohn’s disease or ulcerative colitis
- Hollow organs such as intestine, uterus and spleen prone to rupture
- Developmental delay. A minority of affected individuals have developmental delay. When present, developmental delay is most often associated with craniosynostosis and/or hydrocephalus, suggesting that developmental delay and/or learning disability is an extremely rare primary feature of Loeys-Dietz syndrome. Motor developmental delay is related to muscle hypotonia.
- Less common associated findings of Loeys-Dietz syndrome requiring further exploration include submandibular branchial cysts and defective tooth enamel.
- Pregnancy can be dangerous for women with Loeys-Dietz syndrome due to increased risk of aortic dissection/rupture and uterine rupture
Emergency situations
Following is information regarding potential emergency situations that can arise in someone with Loeys-Dietz Syndrome.
Cardiovascular
Aortic or arterial dissection is a potentially life threatening complication related to aortic aneurysm. Symptoms of aortic dissection include sudden severe chest pain, migrating to the chest, neck, back, abdomen and/or an extremity. Some individuals with aortic dissection have reported nausea, vomiting, shortness of breath, and collapse. Symptoms of aortic dissection warrant calling your local emergency services number and transport to the nearest hospital for aortic imaging (CTA, MRA, echocardiography) to confirm or exclude dissection, for stabilization, and appropriate treatment. Stroke symptoms may present with neck artery dissections.
Pulmonary
Spontaneous pneumothorax results from rupture of pulmonary blebs. Blebs form due to increased air spaces in the lung. Symptoms of spontaneous pneumothorax include chest, neck or back pain exacerbated by deep breathing, or difficult breathing due to pain. Hemoptysis or coughing up blood or blood-tinged mucus from the respiratory tract (lungs and airways) of unknown origin has also been seen in Loeys-Dietz syndrome.
Hollow organ rupture
Hollow organ rupture or tear of the spleen or uterus has been reported rarely. Bowel rupture may be a very rare complication.
Ocular
Retinal detachment is associated with sudden visual field loss. Although retinal detachment is not life threatening, unrecognized/untreated detachment can result in blindness. Therefore, sudden visual disturbance warrants emergency evaluation.
Loeys-Dietz syndrome complications
Several major complications have been noted in early patient studies.
The majority of individuals with Loeys-Dietz syndrome are diagnosed with aneurysms (commonly called enlargements or dilations) of the aortic root. Aneurysms can also be seen throughout the arterial tree (all of the arteries). Although monitoring of aneurysms, surgical intervention and medication use will hopefully help to decrease the amount of cardiovascular complications associated with Loeys-Dietz syndrome, there is a risk for death due to aortic dissection (aneurysm rupture or tear).
Individuals diagnosed with Loeys-Dietz syndrome should go to the emergency room if they have episodes of intense and/or prolonged pain in the body. As many emergency room professionals may not be familiar with the diagnosis of Loeys-Dietz syndrome, it is important that they know of the predisposition to arterial rupture and that prompt imaging should be performed. Rarely, hollow organ (uterus, spleen, intestine) rupture occurs. Individuals impacted by Loeys-Dietz syndrome should work with their physician to develop an emergency letter that should be placed in their medical records and shared with schools, workplaces, etc.
Another complication observed in individuals with Loeys-Dietz syndrome is cervical-spine instability (instability in the vertebrae directly below the skull). This can potentially cause serious risks and should be evaluated with x-rays in the flexion and extension positions.
An orthopedics physician may need to be consulted and may recommend further imaging such as MRI or CT of the neck. These images should be performed prior to any surgeries requiring intubation, as this will impact anesthesia management. A small proportion of individuals with Loeys-Dietz syndrome require surgical intervention to fuse the bones of the skull to prevent dangerous slippage of the bones around the spinal cord.
A complication specific to women is the high incidence of difficulties during pregnancy. There is a high risk of aortic dissection or uterine rupture during pregnancy or directly after childbirth. Many women with Loeys-Dietz syndrome have had numerous successful pregnancies. It is challenging that there are no predictors of which women may experience complications and which may not. If you have a diagnosis of Loeys-Dietz syndrome, you should consult your physicians prior to a pregnancy to discuss risks and to determine a plan for pregnancy management. As some individuals with Loeys-Dietz syndrome are on blood pressure medications in the angiotensin receptor blocker class, it is important to note that this medication is teratogenic (causing medical concerns, birth defects and/or death) to the fetus and a plan for titrating off this medication should be put in place prior to pursuing pregnancy.
Loeys-Dietz syndrome diagnosis
There are several clinical evaluations that are currently pursued together to determine a diagnosis of Loeys-Dietz syndrome.
If there is suspicion of Loeys-Dietz syndrome, it is recommended that individuals be evaluated by a geneticist who is familiar with connective tissue disorders. During the initial visit, a detailed family and medical history will be taken and a comprehensive physical examination will be conducted to evaluate the skeletal, craniofacial and skin-related features that are typically present in individuals with Loeys-Dietz syndrome.
If there is continued suspicion of Loeys-Dietz syndrome, an echocardiogram (ultrasound imaging of the heart) should be performed to assess if there is aortic enlargement and/or other structural heart defects that are consistent with the diagnosis. A consultation with a cardiologist will be necessary to help interpret the cardiac findings.
A physician may also suggest further imaging of the arteries throughout the body. This is done by obtaining a CTA (CT angiogram or computed tomography angiogram) or MRA (MR angiogram or magnetic resonance angiogram) of the entire arterial tree (head, neck, chest, pelvis and abdomen). These imaging studies will detect aneurysms in other arteries.
Three-dimensional reconstruction (3D) of CTA or MRA imaging is recommended to check for arterial tortuosity, a common finding in individuals with Loeys-Dietz syndrome, particularly in the neck. This finding in itself does not typically cause medical concern but can suggest more evidence to support the diagnosis of Loeys-Dietz syndrome.
Genetic testing for mutations (gene changes) within the TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2, TGFB3 and IPO8 genes is clinically available if there is high suspicion of the diagnosis. This test should be ordered by a geneticist, who will be able to accurately interpret and convey the results of the testing. A geneticist will also be able to interpret the family history and determine if genetic testing for other family members is appropriate. If a gene mutation is found in a child, it is typically recommended to test the parents for the same mutation to give accurate recurrence risk information. Testing of offspring of an adult diagnosed with Loeys-Dietz syndrome is always recommended.
Prenatal diagnosis by genetic testing is possible for pregnancies at increased risk for Loeys-Dietz syndrome if the disease-causing gene in the family is already known.
Loeys-Dietz syndrome differential diagnosis
Symptoms of the following disorders may be similar to those of Loeys-Dietz syndrome. Comparisons may be useful for a differential diagnosis.
Marfan syndrome is an autosomal dominant inherited connective tissue disorder with significant clinical overlap with Loeys-Dietz syndrome including aortic root aneurysm, pectus (chest wall) deformities, scoliosis and arachnodactyly. However, individuals with Loeys-Dietz syndrome more often have widely spaced eyes (hypertelorism), cleft palate/bifid uvula, craniosynostosis and/or club feet. Individuals with Loeys-Dietz syndrome also have aneurysms more throughout the arterial tree, where those with Marfan syndrome are most often at the root of the aorta. Individuals with Marfan syndrome are more likely to have eye lens dislocations and heart valve problems.
Individuals diagnosed with Marfan syndrome exhibit several findings not found in Loeys-Dietz syndrome patients. These include:
- Ectopia lentis (dislocation of the lens of the eye)
- Dolichostenomelia (prominently long limbs)
Individuals with Loeys-Dietz syndrome tend to have a more translucent quality to their skin, allowing veins to be easily visible. Abnormal scarring and easy bruising also may occur to a greater degree in individuals with Loeys-Dietz syndrome.
Birth defects such as clubfoot, other structural heart defects and cleft palate (opening and obvious gap in the roof of the mouth) are also more likely to be associated with Loeys-Dietz syndrome.
It has also been discovered that the genetic cause for these two disorders is distinct. Marfan syndrome is caused by genetic mutations (gene change) in the fibrillin-1 (FBN1) gene, while Loeys-Dietz syndrome is caused by a mutation the TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2 or TGFB3 gene.
Shprintzen-Goldberg syndrome is caused by autosomal dominant genetic mutations in the SKI gene, which also interrupts TGF-β activity, similar to Loeys-Dietz syndrome. Symptoms included skeletal and heart abnormalities along with intellectual disability. Intellectual disability is less often seen in Loeys-Dietz syndrome and the skeletal anomalies are usually more profound in Shprintzen-Goldberg syndrome. Individuals with Shprintzen-Goldberg syndrome and oeys-Dietz syndrome may have similar findings, including craniosynostosis, pectus anomalies and scoliosis. However, the vast majority of individuals with Shprintzen-Goldberg syndrome do not show progressive or severe aneurysm formation of the aortic root or of other arteries. Another difference between these two disorders is that individuals with Shprintzen-Goldberg syndrome are also more likely to show developmental delay. Shprintzen-Goldberg syndrome is caused by mutations in the SKI gene.
Vascular Ehlers-Danlos syndrome (vEDS) is an autosomal dominant connective tissue disorder that is also associated with thin skin, aggressive arterial aneurysms, and an increased rate of pregnancy complications such as ruptured uterus. Loeys-Dietz syndrome is similar to vascular type Ehlers-Danlos (vEDS) in that skin-related findings such as easy bruising, soft/velvety skin texture, wide scarring and translucent skin are seen in both syndromes. Both Loeys-Dietz syndrome and vascular type Ehlers-Danlos (vEDS) have a relatively high instance of arterial aneurysms and, to a lesser degree, spontaneous organ rupture. Clubfoot may be observed in both disorders. However, individuals with Loeys-Dietz syndrome have physical findings typically not present in individuals with vascular type Ehlers-Danlos (vEDS) such as widely-spaced eyes and bifid uvula. Vascular type Ehlers-Danlos syndrome (vEDS) is caused by genetic mutations in the COL3A1 gene. Vascular type Ehlers-Danlos syndrome (vEDS) occurs when the collagen an individual produces is not the appropriate quantity or quality. Genetic testing for a mutation in the COL3a1 gene or collagen biochemical studies performed on a skin biopsy sample can confirm this diagnosis. Individuals exhibiting Ehlers-Danlos syndrome-like symptoms but who have had a normal test for vascular type Ehlers-Danlos (vEDS) should be evaluated for Loeys-Dietz syndrome (LDS).
Arterial tortuosity syndrome is an extremely rare autosomal recessive connective tissue disorder caused by genetic mutations of the SLC2A10 gene. Arteries are fragile, twisting, and prone to tearing or narrowing.
Autosomal recessive cutis laxa type 1 also called EFEMP2-related cutis laxa is an autosomal recessive connective tissue disorder characterized by widely spaced eyes, a high arched palate, arterial aneurysms and heart malformations. It is caused by different gene variants than Loeys-Dietz syndrome and does not tend to cause a bifid uvula or skeletal malformations.
Loeys-Dietz syndrome treatment
Loeys-Dietz syndrome manifests itself in a number of ways; therefore, no two persons with Loeys-Dietz syndrome will have identical medical characteristics. It is recommended that you consult your physician or a local geneticist if you have questions about your health concerns. It is always important to consult your doctor to determine an effective and personalized course of action.
There is no known cure for Loeys-Dietz syndrome, but there are treatments directed at specific symptoms. These treatments typically require a team of specialists including a geneticist, cardiologist, heart (cardiothoracic) and bone (orthopedic) surgeons, ophthalmologist, rheumatologist, among others.
Most importantly, the goal of Loeys-Dietz syndrome treatment is to screen and correct blood vessel weaknesses before they tear. Your doctor may prescribe a beta-blocker or angiotensin receptor blocker (ARB) such as losartan to slow down the ballooning or stretching of the aortic root. Every 6 months to 1 year, you should be screened for aneurysms of the entire arterial tree from your head to your hips. This screening includes blood vessel imaging by CT angiography (CTA) or MR angiography (MRA) and heart imaging by echocardiography. If repeated imaging of the entire arterial tree does not show any changes or concern for aneurysms, it may be acceptable to image less frequently. Your doctor will be looking to see whether the aortic root balloons to larger than 4 cm (1.5 inches), at which point he/she may recommend surgery to replace the ballooning section of the aorta and sometimes the aortic valve of the heart. This surgery is typically safe and effective in fixing the problem.
To treat some of the musculoskeletal abnormalities associated with Loeys-Dietz syndrome, your care team may recommend nonsurgical management of the symptoms such as bracing or surgical correction of the abnormalities. These abnormalities include an abnormally curved spine (scoliosis), an indented or protruding chest, and issues with the bones of the neck. Doctors will recommend X-rays and CT scans to determine whether surgery should be performed. These surgeries can be complicated and require close attention for complications afterwards.
Staying active and conditioned is recommended for individuals with Loeys-Dietz syndrome. Athletic goals and limits should be discussed with the cardiologist and the care team to determine an individualized health plan.
Table 2. Loeys-Dietz Syndrome Recommended Evaluations Following Diagnosis
| System or Concern | Evaluation | Comment |
|---|---|---|
| Cardiovascular |
| Aortic root measurements must be interpreted based on consideration of normal values for age and body size. Approximately half of persons with Loeys-Dietz syndrome studied had an aneurysm distant from the aortic root that would not have been detected by echocardiography 19 |
| Skeletal |
| |
| Craniofacial |
| |
| Allergy / Gastrointestinal disease |
| |
| Ocular |
| |
| Genetic counseling | By genetics professionals | To obtain a pedigree & inform affected persons and their families re nature, mode of inheritence & implications of Loeys-Dietz syndrome to facilitate medical & personal decision making |
Table 3. Loeys-Dietz syndrome treatment options
| Manifestation or Concern | Treatment | Considerations/Other |
|---|---|---|
| Cardiovascular | Persons should be managed in a medical center familiar with Loeys-Dietz syndrome. | |
| Beta-blockers or angiotensin receptor blockers (ARBs) to reduce hemodynamic stress | No clinical trials evaluating efficacy of beta-blockers vs angiotensin receptor blockers (ARBs) have been completed in persons with Loeys-Dietz syndrome. | |
Aneurysms are amenable to early and aggressive surgical intervention:
Note: Suggested surgical thresholds vary based on involved gene, family history, aortic root growth rate, and valve function, among other variables. Consider subacute bacterial endocarditis prophylaxis in persons w/mitral &/or aortic regurgitation who are undergoing dental work or other procedures expected to contaminate the bloodstream w/bacteria. |
| |
| Pectus excavatum | Rarely, surgical intervention is medically (rather than cosmetically) indicated. | |
| Congenital hip dislocation / Other joint manifestations | Management per orthopedist | |
| Clubfeet | Surgical correction by orthopedic surgeon | |
| Cervical spine instability | Surgical fixation may be necessary to prevent damage to spinal cord. | Because of high risk of cervical spine instability, cervical spine flexion & extension radiographs should be performed prior to intubation or any other procedure involving manipulation of neck. |
| Spondylolisthesis | Mgmt per orthopedist | |
| Scoliosis | Progressive scoliosis should be managed by orthopedist; surgical stabilization of spine may be required. | |
| Acetabular protrusion | Surgical intervention is rarely indicated; treatment focuses on pain control. | |
| Pes planus | Orthotics are only indicated for severe pes planus. Some persons prefer use of arch supports, while others find them irritating; the choice should be left to personal preference. | Surgical intervention is rarely indicated or successful. |
| Fracture/Osteoporosis | Severe osteoporosis w/pathologic fracture may require specific treatment under guidance of endocrinologist. 1 | |
| Cleft palate / Craniosynostosis | Standard mgmt by craniofacial team | |
| Hernias | A supporting mesh can be used during surgical repair to minimize recurrence risk. | Hernias tend to recur after surgical intervention. |
| Allergic/ Inflammatory |
| |
| Ocular |
| |
| Pneumothorax | Optimal mgmt of pneumothorax to prevent recurrence may require chemical or surgical pleurodesis or surgical removal of pulmonary blebs. | |
| Organ rupture | Counseling regarding other life-threatening manifestations incl spontaneous rupture of spleen & bowel & pregnancy-assoc risks is recommended. | |
| Dural ectasia | Dural ectasia is usually asymptomatic; no effective therapies for symptomatic dural ectasia currently exist. |
Medications
Pressure on the aorta (the largest artery leaving the heart) and other arteries can be controlled by the administration of medications that work to lessen the strain on the body’s major arteries by reducing heart rate and blood pressure. In mouse models of Loeys-Dietz syndrome the specific class of blood-pressure lowering medications known as angiotensin receptor blockers (ARBs), has shown great benefit in reducing aneurysm growth. In people impacted by Loeys-Dietz syndrome, if this type of medication is used, it should be used at optimal titration. Angiotensin receptor blockers include medications by the name of Losartan, Irbesartan or Candesartan. Many people are also maintained on beta-blockers (Atenolol, Metoprolol). It is recommended that individuals with Loeys-Dietz syndrome remain on these medications even after surgical repair of aneurysms.
Imaging
Continued monitoring of the aorta through annual echocardiograms is necessary. A baseline CT angiography (CTA) or MR angiography (MRA) of the head, neck, chest, abdomen and pelvis should also be performed to detect and monitor aneurysm formation and/or dissections (tears). The frequency of these scans depends on aneurysm size and rate of growth, so it is recommended that individuals consult their doctor on the appropriate intervals for imaging. Individuals should try not to go more than two years without head-to-pelvis imaging. If a person is using MR angiography (MRA) imaging for surveillance, every few imaging cycles a CT angiography (CTA) of the head and neck should be considered, as this imaging has better clarity of small arteries in the head and neck.
If repeated imaging of the entire arterial tree does not show any changes or concern for aneurysms, it may be acceptable to image less frequently. Your doctor will be looking to see whether the aortic root balloons to larger than 4 cm (1.5 inches), at which point he/she may recommend surgery to replace the ballooning section of the aorta and sometimes the aortic valve of the heart. This surgery is typically safe and effective in fixing the problem.
Cervical Spine Imaging: X-rays of the cervical spine in the flexion and extension positions are recommended to assess for vertebral anomalies and/or instability. If there are any anomalies detected, consultation with an orthopedist is recommended. Rarely, surgery for cervical spine fusion is required. It is important to assess for cervical spine instability prior to undergoing any surgery, as this may impact intubation procedures.
Blood Vessel Surgery
Vascular surgery is a widely recommended treatment option as a preventative surgery for individuals with a rapidly enlarging aorta or artery or a pronounced family history of arterial dissection. Aortic root replacement is the most common vascular surgery occurring in individuals with Loeys-Dietz syndrome, and it is highly successful. There are many examples of successful arterial repairs for aneurysms throughout the body through a variety of surgical interventions as vascular tissue is not typically weak or fragile in individuals with Loeys-Dietz syndrome.
Exercise Restrictions
Exercise restrictions are typically put in place to assist in slowing the rate of aortic and arterial aneurysm growth. It is advised that individuals with Loeys-Dietz syndrome avoid competitive sports, especially contact sports, and other exercises or muscle straining activities performed to the point of exhaustion. Individuals can and should remain active with aerobic types of activities that are performed in moderation. Exercises where you have to strain your muscles, such as push-ups, chin-ups, sit-ups, are to be avoided. Activities such as hiking, biking, jogging and swimming that help to naturally lower the heart rate and blood pressure should be a part of an individual’s cardiovascular activity. A good recommendation for cardiovascular activities is to exercise only to a level where you can hold a conversation while performing the activity.
Orthopedics
People with Loeys-Dietz syndrome have musculoskeletal abnormalities that include an abnormally curved spine (scoliosis), an indented or protruding chest, and issues with the bones of the neck. Doctors will recommend X-rays and CT scans to determine whether surgery should be performed. To treat some of these musculoskeletal abnormalities, your care team may recommend nonsurgical management of the symptoms such as bracing for scoliosis, orthotics/surgeries for foot deformities or contractures or harnesses for congenital hip dislocation. These surgeries can be complicated and require close attention for complications afterwards. Typically, surgery for pectus anomalies is pursued for cosmetic purposes and not out of medical necessity.
Allergies
Environmental and food allergies are increased in individuals with Loeys-Dietz syndrome and may require a consultation with an allergist or gastroenterologist. Allergic reactions may present as rhinitis or sinusitis, eczema, or hives. Gastrointestinal complaints can include the feeling of food getting stuck in your throat, diarrhea, abdominal pain or difficulty gaining weight. Some individuals have severe inflammatory disease of the esophagus or intestines that may need stricter intervention such as medications or feeding tubes to help with caloric intake.
Drugs and circumstances to avoid
Drugs or medications that stimulate the cardiovascular system including routine use of decongestants or triptan medications for the management of migraine headache should be avoided.
For individuals at risk for recurrent pneumothorax, breathing against a resistance (e.g., playing a brass instrument) or positive pressure ventilation (e.g., scuba diving) should be avoided.
Pregnancy management
Pregnancy and the postpartum period can be dangerous for women with Loeys-Dietz syndrome because of increased risk of aortic dissection/rupture and uterine rupture. Increased frequency of aortic imaging is recommended, both during pregnancy and in the weeks following delivery.
Loeys-Dietz syndrome life expectancy
The major sources of morbidity and early mortality in Loeys-Dietz syndrome are dilatation of the aorta at the level of the sinuses of Valsalva, a predisposition for aortic dissection and rupture, mitral valve prolapse with or without regurgitation, and enlargement of the proximal pulmonary artery 5.
Individuals with Loeys-Dietz syndrome have a more aggressive vascular course (with routine involvement of vascular segments distant from the aortic root) than that observed in Marfan syndrome. Mean age at death is 26 years 20. Attias et al 21 reported that the proportion of individuals with aortic dilatation, the age at dissection, and the need for surgery were similar in those with a heterozygous TGFBR2 pathogenic variant and those with a heterozygous FBN1 pathogenic variant causative of Marfan syndrome; however, the rate of death was greater in families with a heterozygous TGFBR2 pathogenic variant. Similarly, a study of 228 families with a heterozygous pathogenic variant in either TGFBR1 or TGFBR2 demonstrated similar aortic risk (dissection or aortic surgery) in both groups 22.
- Loeys BL, Dietz HC. Loeys-Dietz Syndrome. 2008 Feb 28 [Updated 2024 Sep 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1133[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- About Loeys-Dietz Syndrome. https://www.loeysdietz.org/en/medical-information[↩][↩][↩]
- Loeys-Dietz Syndrome. https://rarediseases.org/rare-diseases/loeys-dietz-syndrome[↩][↩][↩]
- Ziegler A, Duclaux-Loras R, Revenu C, et al. Bi-allelic variants in IPO8 cause a connective tissue disorder associated with cardiovascular defects, skeletal abnormalities, and immune dysregulation. Am J Hum Genet. 2021 Jun 3;108(6):1126-1137. doi: 10.1016/j.ajhg.2021.04.020[↩]
- Loeys BL, Dietz HC. Loeys-Dietz Syndrome. 2008 Feb 28 [Updated 2018 Mar 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1133[↩][↩][↩]
- Lynch CP, Patel M, Seeley AH, Seeley MA. Orthopaedic Management of Loeys-Dietz Syndrome: A Systematic Review. J Am Acad Orthop Surg Glob Res Rev. 2021 Nov 15;5(11):e21.00087. doi: 10.5435/JAAOSGlobal-D-21-00087[↩]
- Baldo F, Morra L, Feresin A, Faletra F, Al Naber Y, Memo L, Travan L. Neonatal presentation of Loeys-Dietz syndrome: two case reports and review of the literature. Ital J Pediatr. 2022 Jun 6;48(1):85. doi: 10.1186/s13052-022-01281-y[↩]
- Loeys BL, Chen J, Neptune ER, Judge DP, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005 Mar;37(3):275-81. doi: 10.1038/ng1511[↩]
- Loeys BL, Schwarze U, Holm T, Callewaert BL, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006 Aug 24;355(8):788-98. doi: 10.1056/NEJMoa055695[↩]
- Granadillo JL, Chung WK, Hecht L, Corsten-Janssen N, Wegner D, Nij Bijvank SWA, Toler TL, Pineda-Alvarez DE, Douglas G, Murphy JJ, Shimony J, Shinawi M. Variable cardiovascular phenotypes associated with SMAD2 pathogenic variants. Hum Mutat. 2018 Dec;39(12):1875-1884. doi: 10.1002/humu.23627[↩]
- Cannaerts E, Kempers M, Maugeri A, Marcelis C, Gardeitchik T, Richer J, Micha D, Beauchesne L, Timmermans J, Vermeersch P, Meyten N, Chénier S, van de Beek G, Peeters N, Alaerts M, Schepers D, Van Laer L, Verstraeten A, Loeys B. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: further delineation of the phenotype. J Med Genet. 2019 Apr;56(4):220-227. doi: 10.1136/jmedgenet-2018-105304[↩]
- Wischmeijer A, Van Laer L, Tortora G, et al. Thoracic aortic aneurysm in infancy in aneurysms-osteoarthritis syndrome due to a novel SMAD3 mutation: further delineation of the phenotype. Am J Med Genet A. 2013 May;161A(5):1028-35. doi: 10.1002/ajmg.a.35852[↩]
- van de Laar IM, Oldenburg RA, Pals G, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011 Feb;43(2):121-6. doi: 10.1038/ng.744[↩]
- Boileau C, Guo DC, Hanna N, Regalado ES, et al. National Heart, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project; Jondeau G, Milewicz DM. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012 Jul 8;44(8):916-21. doi: 10.1038/ng.2348[↩][↩]
- Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012 Jul 8;44(8):922-7. doi: 10.1038/ng.2349[↩][↩]
- Bertoli-Avella AM, Gillis E, Morisaki H, et al. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015 Apr 7;65(13):1324-1336. doi: 10.1016/j.jacc.2015.01.040[↩][↩]
- Van Gucht I, Meester JAN, Bento JR, et al. A human importin-β-related disorder: Syndromic thoracic aortic aneurysm caused by bi-allelic loss-of-function variants in IPO8. Am J Hum Genet. 2021 Jun 3;108(6):1115-1125. doi: 10.1016/j.ajhg.2021.04.019[↩]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81[↩]
- Roman MJ, Devereux RB, Kramer-Fox R, O’Loughlin J. Two-dimensional echocardiographic aortic root dimensions in normal children and adults. Am J Cardiol. 1989 Sep 1;64(8):507-12. doi: 10.1016/0002-9149(89)90430-x[↩]
- Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–98[↩]
- Attias D, Stheneur C, Roy C, Collod-Béroud G, Detaint D, Faivre L, Delrue MA, Cohen L, Francannet C, Béroud C, Claustres M, Iserin F, Khau Van Kien P, Lacombe D, Le Merrer M, Lyonnet S, Odent S, Plauchu H, Rio M, Rossi A, Sidi D, Steg PG, Ravaud P, Boileau C, Jondeau G. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120:2541–9[↩]
- Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, De Backer J, Muiño-Mosquera L, Naudion S, Zordan C, Morisaki T, Morisaki H, Von Kodolitsch Y, Dupuis-Girod S, Morris SA, Jeremy R, Odent S, Adès LC, Bakshi M, Holman K, LeMaire S, Milleron O, Langeois M, Spentchian M, Aubart M, Boileau C, Pyeritz R, Milewicz DM., Montalcino Aortic Consortium. International registry of patients carrying TGFBR1 or TGFBR2 mutations: results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet. 2016;9:548–58[↩]


{kind=link}