
Contents
What is Marfan syndrome
Marfan syndrome (MFS) also known as Marfan’s syndrome is a inherited genetic disorder that affects the connective tissue in many parts of your body 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17. Connective tissue provides strength and flexibility to structures such as bones, ligaments, muscles, blood vessels, skin, lungs and heart valves. Marfan syndrome generally affects the limbs, but can also affect the spine, sternum, eyes, heart and blood vessels. The signs and symptoms of Marfan syndrome vary widely in severity, timing of onset, and rate of progression. Because connective tissue is found throughout your body, Marfan syndrome can affect many parts of your body such as your heart and blood vessels (cardiovascular), skeletal (bones, and joints) and eye (ocular) systems. The 2 main features of Marfan syndrome are vision problems caused by a dislocated lens in one or both eyes (ectopia lentis) and defects in the large blood vessel that distributes blood from the heart to the rest of the body called the aorta. The aorta can weaken and stretch, which may lead to a bulge in the blood vessel wall (aortic aneurysm). Stretching of the aorta may cause the aortic valve to leak, which can lead to a sudden tearing of the layers in the aorta wall (aortic dissection). Approximately 80% of patients will develop aortic complications. Aortic aneurysm and aortic dissection can be life threatening and is the cause of death in 30-45% of individuals with Marfan’s syndrome. Many people with Marfan syndrome have additional heart problems including a leak in the valve that connects two of the four chambers of the heart (mitral valve prolapse) or the valve that regulates blood flow from the heart into the aorta (aortic valve regurgitation). Leaks in these heart valves can cause shortness of breath, fatigue, and an irregular heartbeat felt as skipped or extra beats (palpitations).
Individuals with Marfan syndrome are usually tall and slender and have an arm span that exceeds their body height due to an overgrowth of the long bones of the arms and legs. Individuals with Marfan syndrome also have abnormally long slender fingers and toes resembling a spider’s legs (arachnodactyly), loose joints (joint hypermobility), long and narrow face, crowded teeth, abnormal side-to-side curvature of the spine (scoliosis) or forward curvature of the spine resulting in a “hunchback” or rounded appearance (kyphosis), stretch marks (striae) not related to weight gain or loss, and either a sunken chest (pectus excavatum) or a protruding chest (pectus carinatum). Some individuals develop an abnormal accumulation of air in the chest cavity that can result in the collapse of a lung (spontaneous pneumothorax). A membrane called the dura, which surrounds the brain and spinal cord, can be abnormally enlarged (dural ectasia) in people with Marfan syndrome. Dural ectasia can cause pain in the back, abdomen, legs, or head. Most individuals with Marfan syndrome have some degree of nearsightedness (myopia). Clouding of the lens of the eye (cataract) may occur in mid-adulthood, and increased pressure within the eye (glaucoma) occurs more frequently in people with Marfan syndrome than in those without the condition. Marfan’s syndrome does not affect intelligence.
Note that these signs and symptoms and the severity of Marfan syndrome vary greatly from person to person. The features of Marfan syndrome can become apparent anytime between infancy and adulthood. Patients may be diagnosed with Marfan’s syndrome at birth, during childhood or later in life. The more severe cases are usually diagnosed early. Many patients present with features such as tall stature and an arm span that exceeds height. However, eye lens dislocation, aortic dilatation or skeletal signs and symptoms such as chest deformities (pectus excavatum or pectus carinatum) or curvature of the spine (scoliosis or kyphosis) may also be the first presentation. Many of the signs and symptoms of Marfan’s syndrome change with age so diagnosis can be difficult in children.
Depending on the onset and severity of signs and symptoms, Marfan syndrome can be fatal early in life; however, with proper treatment, many affected individuals have normal lifespans surviving into mid- to late adulthood.
If you have Marfan syndrome, it is important to understand how it affects your body and to be aware of your physical limitations. You can help prevent unnecessary stress or strain on joints by taking care during physical activity.
Marfan syndrome treatment usually includes medications to lower your blood pressure to reduce the strain on your aorta. Some people may need annual check-ups to monitor the heart. Regular monitoring to check for damage progression is vital. Many people with Marfan syndrome eventually require preventive surgery to repair damaged heart valves, blood vessels and joints.
Marfan syndrome treatment guidelines for all adults 2, 18:
- Restriction of physical activity with avoidance of contact sports, isometric exercise, and activities that can cause joint injury/pain
- Avoidance of agents that stimulate the cardiovascular system, such as decongestants and caffeine
- Annual ophthalmologic examination – LASIK correction of refractive errors is not recommended.
- Subacute endocarditis prophylaxis for dental work in the presence of mitral or aortic valve regurgitation
- Annual echocardiography to evaluate the ascending aorta in cases of small aortic dimensions and/or a slow aortic dilatation rate – Echocardiography is required with greater frequency when aortic root diameter is over 4.5 cm in adults, if aortic dilatation is greater than 0.5 cm per year, and if significant aortic regurgitation is present
- Beta-blockers or angiotensin receptor blockers (ARBs) treatment to reduce hemodynamic stress on the aortic wall. Current American College of Cardiology/American Heart Association guidelines recommend use of either beta blockers or angiotensin receptor blockers (ARBs) or a combination of both at maximally tolerated doses 19. European guidelines recommend use of beta blockers and consider angiotensin receptor blockers (ARBs) as an alternative treatment 20.
Marfan syndrome is caused by changes or mutations (variants) in the fibrillin-1 (FBN1) gene in the long arm of chromosome 15 at position 21.1 (15q21.1) 21.
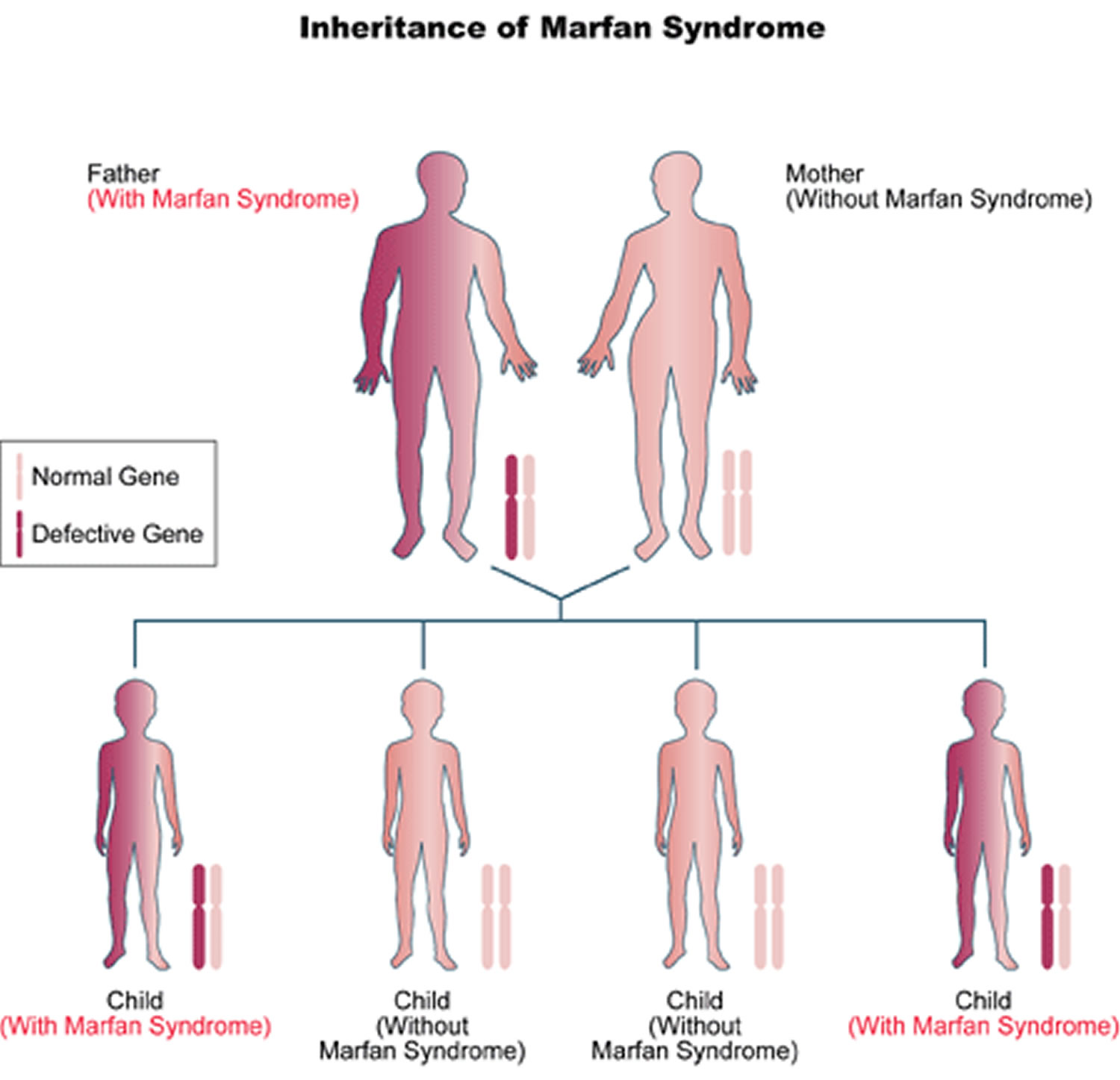
About 3 out of 4 people (75% of patients) with Marfan syndrome inherit it, meaning they get the fibrillin-1 (FBN1) genetic mutation from a parent who has it. Most Marfan syndrome follows an autosomal dominant inheritance pattern, meaning that only one copy of a FBN1 gene variant is sufficient to cause Marfan syndrome. Autosomal dominant refers to a pattern of genetic inheritance where a genetic trait or condition is passed on when only one copy of a mutated gene (FBN1 gene) is inherited from a single parent. “Autosomal” means the gene is located on one of the numbered chromosomes (not sex chromosomes), and “dominant” means the altered gene is expressed even when a working copy is also present. People with an autosomal dominant condition have a 50% chance of passing the altered gene to each child. Meaning there is a 50 percent chance that a person with Marfan syndrome will pass along the genetic mutation each time they have a child. A child with a parent who has the mutated FBN1 gene has a 50% chance of inheriting Marfan syndrome. Both males and females are equally likely to inherit and be affected by autosomal dominant conditions. However, there are rare case reports describing autosomal recessive fibrillin 1 gene (FBN1) mutations 22. In autosomal recessive pattern of genetic inheritance, Marfan syndrome only appears only in individuals who have inherited 2 copies of a mutated or altered FBN1 gene, one from each parent, located on a non-sex (autosomal) chromosome. Both parents of an affected individual are typically “carriers,” possessing one mutated copy and one working copy of the FBN1 gene, but do not themselves show symptoms of Marfan syndrome. In autosomal recessive inheritance if a child inherits one mutated FBN1 gene and one working copy, the child will be a carrier but will not have Marfan syndrome.
About 25% of patients with Marfan syndrome are the first in their family to have it, when this happens it is called a spontaneous mutation or “de novo mutation” 23, 24. These spontaneous mutation or “de novo mutation” Marfan syndrome cases are more severely affected. A de novo mutation is a genetic alteration that appears for the first time in an individual and is not inherited from either parent. These mutations can occur in a parent’s egg or sperm cell, or in the fertilized egg itself during early development, leading to a new genetic change in that person. De novo mutations are responsible for some genetic disorders and diseases, as they represent fresh genetic changes that were not present in the prior generations of a family.
In less than 10% of patients with typical Marfan phenotype, no mutation in FBN1 is identifiable, likely due to complete allele deletion or altered regulation of the FBN1 gene 2. In patients with atypical presentations reminiscent of Marfan syndrome, a mutation in a gene encoding for transforming growth factor-beta receptor (TGFBR) may be the cause 2. Some individuals with TGFBR1 (Transforming Growth Factor Beta Receptor 1) or TGFBR1 (Transforming Growth Factor Beta Receptor 2) gene mutations have clinical features consistent with Marfan syndrome, while others have features of 1 of 2 other syndromes such as Loeys-Dietz syndrome (LDS) or familial thoracic aortic aneurysm (FTAA) syndrome 1.
The FBN1 gene provides instructions for making a protein called fibrillin-1 25. Fibrillin-1 attaches (binds) to other fibrillin-1 proteins and other molecules to form threadlike filaments called microfibrils 3. Microfibrils become part of the fibers that provide strength and flexibility to connective tissue 3. Additionally, microfibrils bind to molecules called growth factors and release them at various times to control the growth and repair of tissues and organs throughout the body 3. A mutation in the FBN1 gene can reduce the amount of functional fibrillin-1 that is available to form microfibrils, which leads to decreased microfibril formation 26, 27. As a result, microfibrils cannot bind to growth factors, so excess growth factors are available and elasticity in many tissues is decreased, leading to overgrowth and instability of tissues in Marfan syndrome 3.
Approximately 1 per 3,000 to 1 per 5,000 individuals is affected by Marfan syndrome 28 Marfan syndrome occurs worldwide, with no preference for race or gender. Marfan syndrome exhibits complete penetrance with variable expression 29.
Figure 1. The anatomy of the heart valves
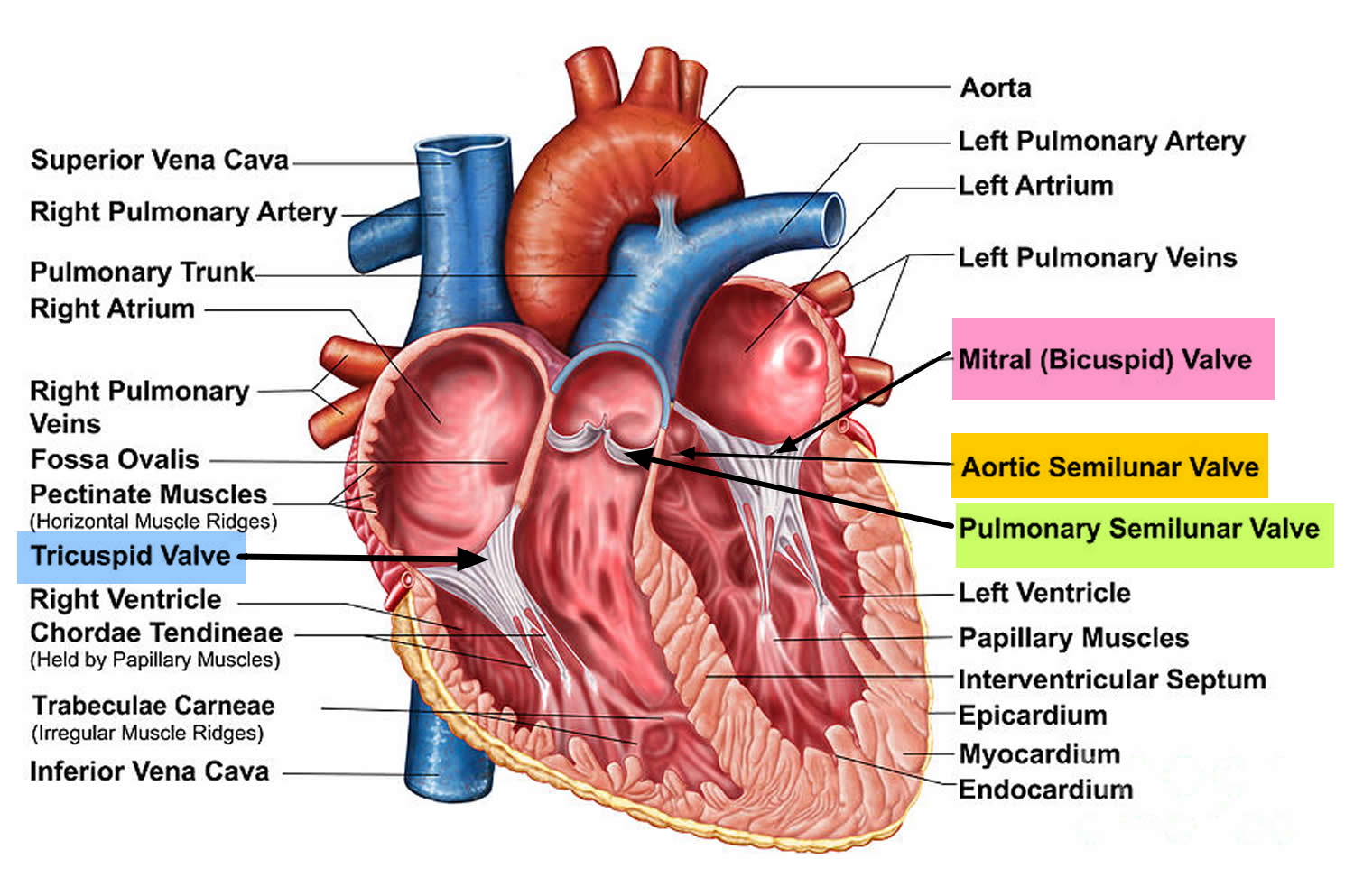
Figure 2. Heart valves function
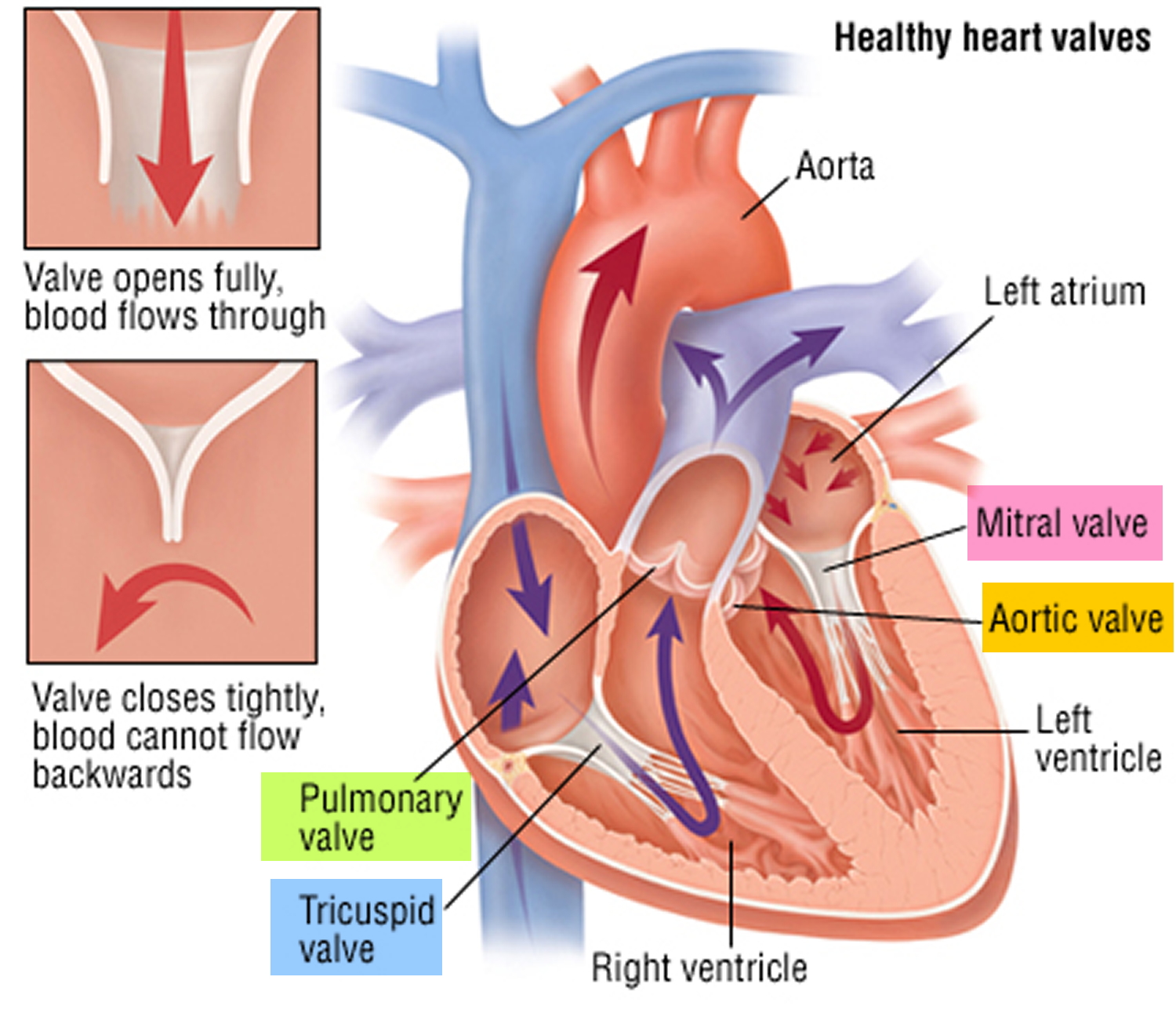
Figure 3. Arachnodactyly
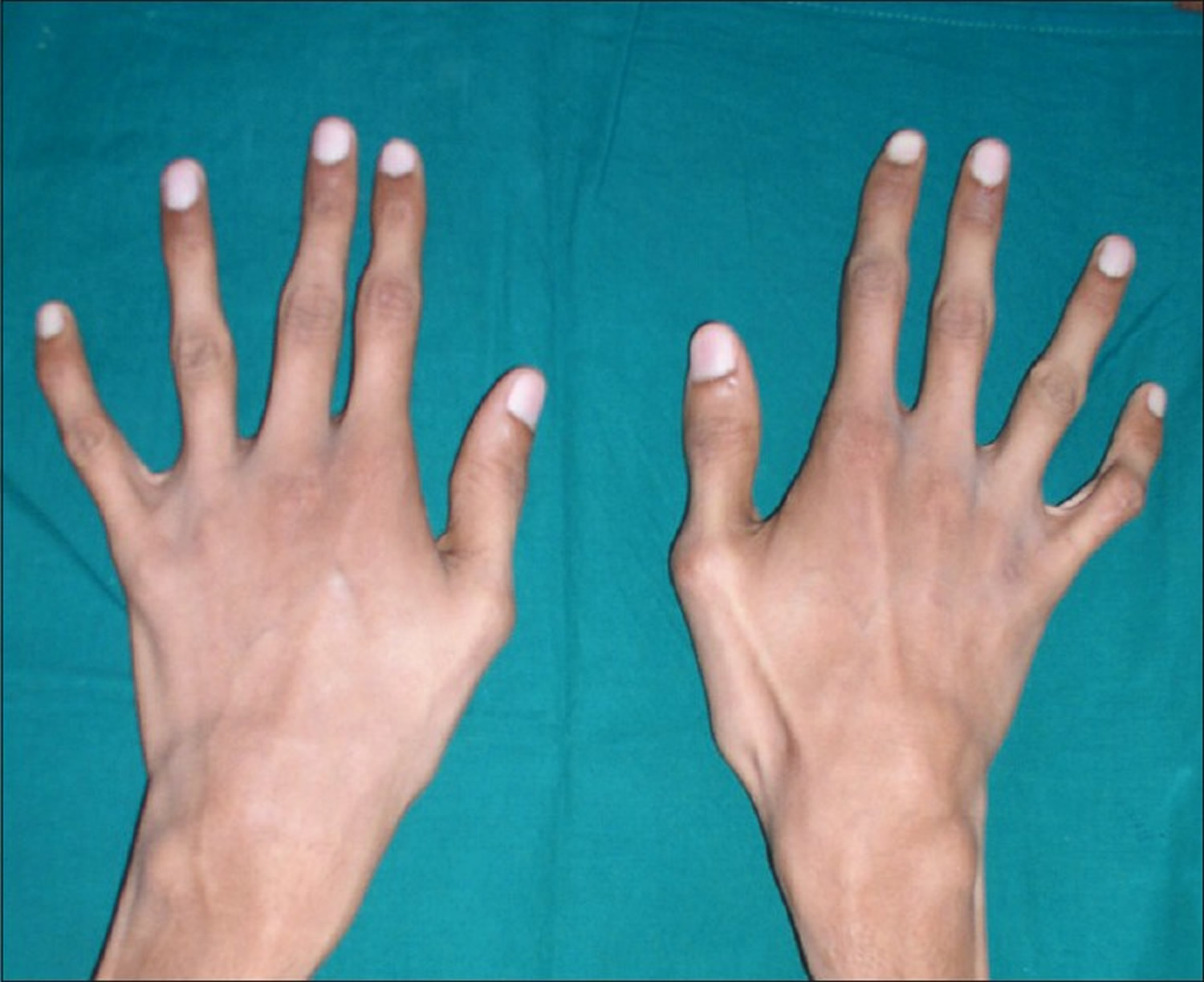
Figure 4. Marfan syndrome abnormal curvature of the spine (scoliosis or kyphosis)
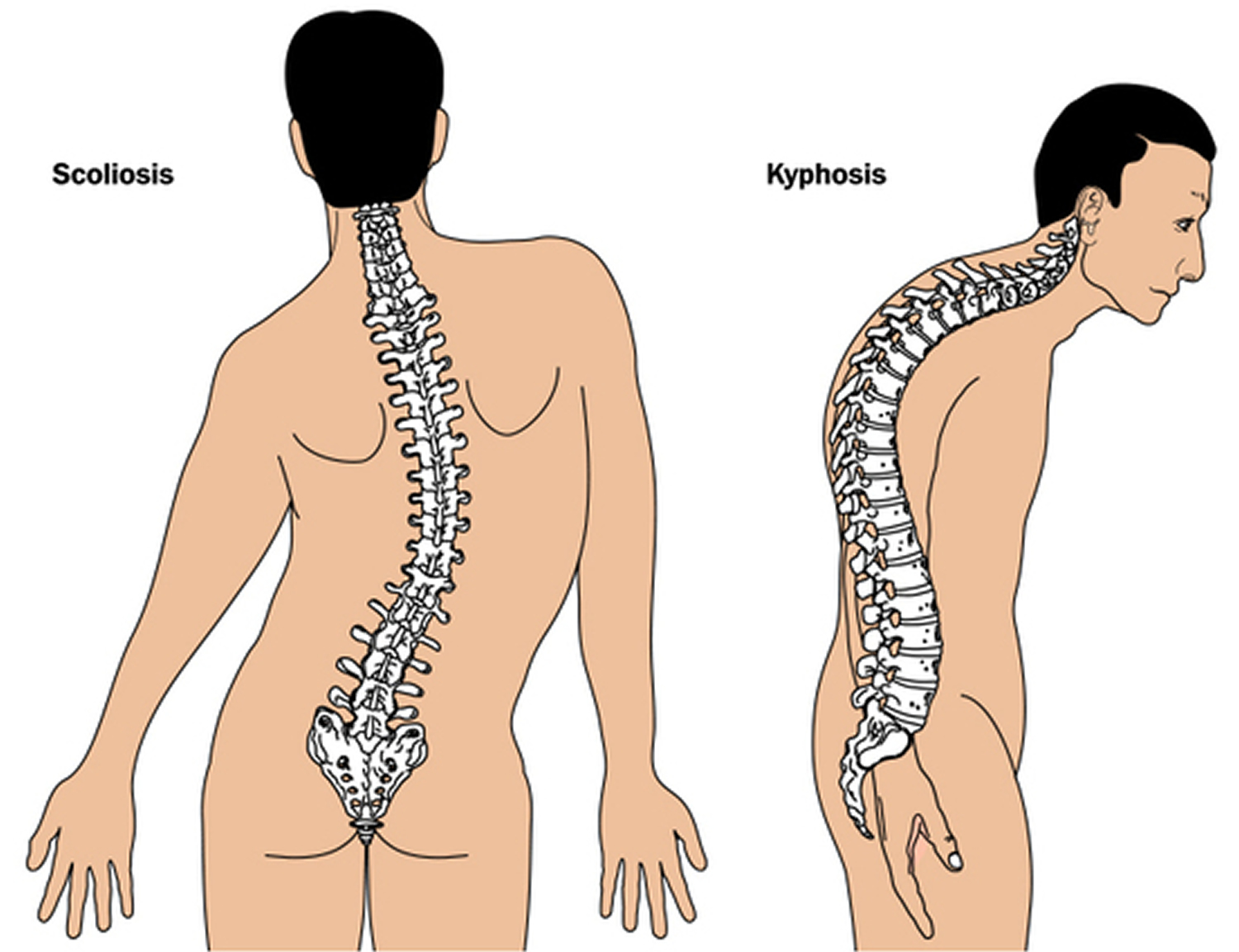
Figure 5. Marfan syndrome – sunken chest (pectus excavatum)
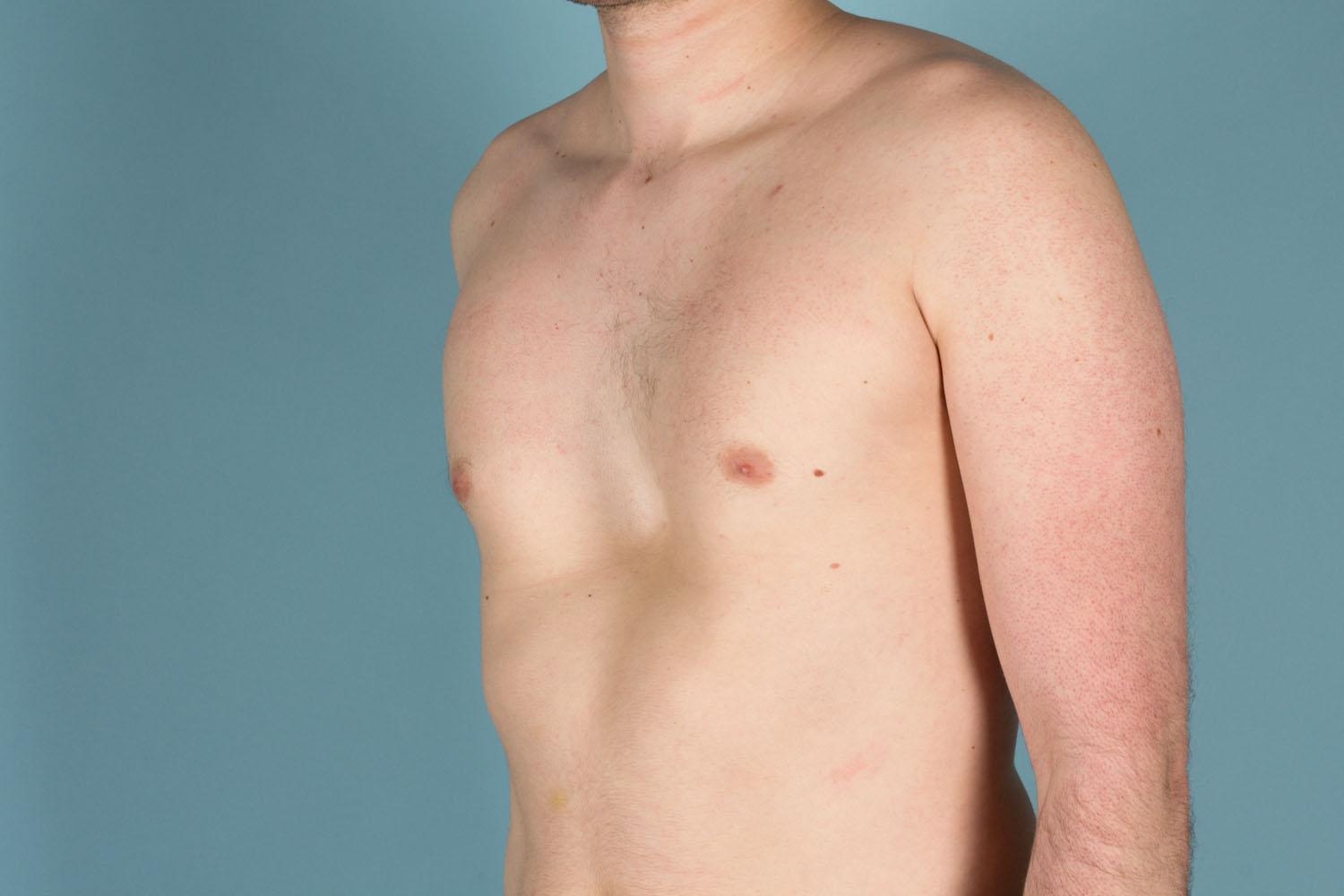
Figure 6. Marfan syndrome – protruding chest (pectus carinatum)
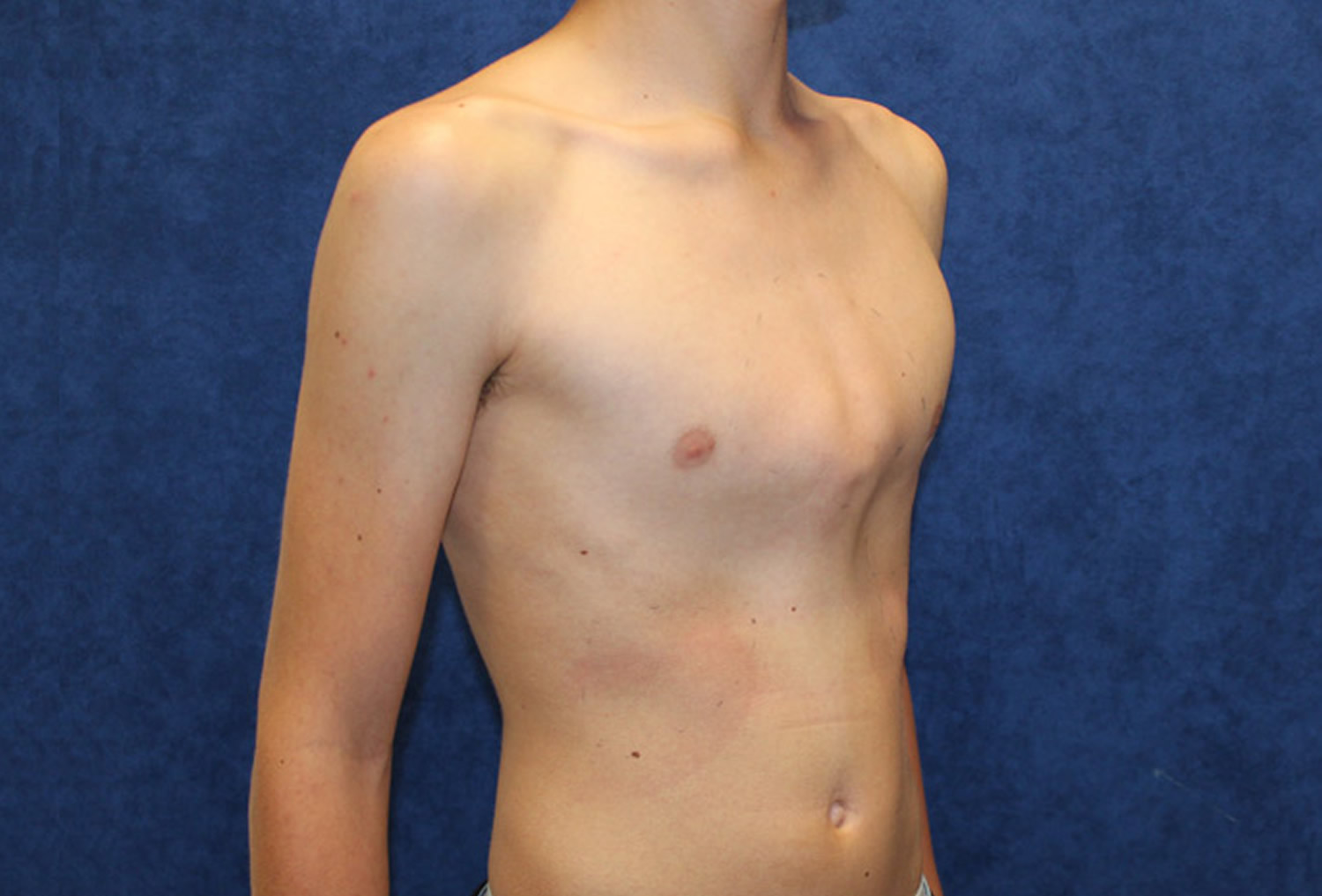
Figure 7. Marfan syndrome arm span that exceeds their body height

Marfan syndrome causes
Over 90% of patients Marfan syndrome is caused by changes or mutations (variants) in the fibrillin-1 (FBN1) gene in the long arm of chromosome 15 at position 21.1 (15q21.1) that affects the elasticity of connective tissue in muscles and joints. The FBN1 gene provides instructions for making a large protein called fibrillin-1 25. The fibrillin-1 protein is transported out of cells into the extracellular matrix, which is an intricate lattice of proteins and other molecules that forms in the spaces between cells. In this matrix, molecules of fibrillin-1 attach (bind) to each other and to other proteins to form threadlike filaments called microfibrils 26, 27. Microfibrils form elastic fibers, which enable the skin, ligaments, and blood vessels to stretch. Microfibrils also provide strength and flexibility to connective tissues such as bones and the tissues that support the nerves, muscles, and lenses of the eyes. Microfibrils store a protein called transforming growth factor beta (TGF-β), a critical growth factor. Transforming growth factor beta (TGF-β) affects development by helping to control the growth and division (proliferation) of cells, the process by which cells mature to carry out specific functions (differentiation), cell movement (motility), and the self-destruction of cells (apoptosis). Microfibrils help regulate the availability of transforming growth factor beta (TGF-β), which is turned off (inactivated) when stored in microfibrils and turned on (activated) when released. A mutation in the FBN1 gene can reduce the amount of functional fibrillin-1 that is available to form microfibrils, which leads to decreased microfibril formation. As a result, microfibrils cannot bind to transforming growth factor beta (TGF-β), so excess TGF-β growth factors are available and elasticity in many tissues is decreased, leading to overgrowth and instability of tissues in Marfan syndrome 3.
The role of transforming growth factor beta (TGF-β) in the pathophysiology of Marfan syndrome has been solidified by the use of the therapeutic angiotensin-converting enzyme inhibitors (ACE inhibitors) and angiotensin 2 receptor blockers (ARBs), both proven to decrease TGF-beta activity 2. Early studies in a mouse model of Marfan syndrome have shown the utility of a TGF-beta-neutralizing antibody or angiotensin receptor blocker (ARB) losartan in treating the disease 30. Comparatively, mutant mice treated with a beta blocker propranolol only exhibited a moderate reduction in the rate of aortic root dilation. Human studies substantiated that angiotensin receptor blocker (ARB) therapy significantly reduced the rate of change in the aortic root diameter compared to beta-blocker therapy alone 31.
- Marfan syndrome is most often inherited from a parent in an autosomal dominant pattern, who will have a 50% chance of passing the condition on to their children.
- However, in about one quarter of people diagnosed with Marfan syndrome, nobody else in the family is affected – the disease is due to a new mutation in the FBN1 gene 32, 33. These cases occur in people with no history of the disorder in their family. These spontaneous mutation or “de novo mutation” Marfan syndrome cases are more severely affected.
- There are rare case reports describing autosomal recessive fibrillin 1 gene (FBN1) mutations 22. In autosomal recessive pattern of genetic inheritance, Marfan syndrome only appears only in individuals who have inherited 2 copies of a mutated or altered FBN1 gene, one from each parent, located on a non-sex (autosomal) chromosome. Both parents of an affected individual are typically “carriers,” possessing one mutated copy and one working copy of the FBN1 gene, but do not themselves show symptoms of Marfan syndrome. In autosomal recessive inheritance if a child inherits one mutated FBN1 gene and one working copy, the child will be a carrier but will not have Marfan syndrome.
In less than 10% of patients with typical Marfan phenotype, no mutation in FBN1 is identifiable, likely due to complete allele deletion or altered regulation of the FBN1 gene 2. In patients with atypical presentations reminiscent of Marfan syndrome, a mutation in a gene encoding for transforming growth factor-beta receptor (TGFBR) may be the cause 2. Some individuals with TGFBR1 (Transforming Growth Factor Beta Receptor 1) or TGFBR1 (Transforming Growth Factor Beta Receptor 2) gene mutations have clinical features consistent with Marfan syndrome, while others have features of 1 of 2 other syndromes such as Loeys-Dietz syndrome (LDS) or familial thoracic aortic aneurysm (FTAA) syndrome 1.
How is marfan syndrome inherited?
About 3 out of 4 people (75% of patients) with Marfan syndrome inherit it, meaning they get the fibrillin-1 (FBN1) genetic mutation from a parent who has it. Most Marfan syndrome follows an autosomal dominant inheritance pattern, meaning that only one copy of a FBN1 gene variant is sufficient to cause Marfan syndrome. Autosomal dominant refers to a pattern of genetic inheritance where a genetic trait or condition is passed on when only one copy of a mutated gene (FBN1 gene) is inherited from a single parent. “Autosomal” means the gene is located on one of the numbered chromosomes (not sex chromosomes), and “dominant” means the altered gene is expressed even when a working copy is also present. People with an autosomal dominant condition have a 50% chance of passing the altered gene to each child. Meaning there is a 50 percent chance that a person with Marfan syndrome will pass along the genetic mutation each time they have a child. A child with a parent who has the mutated FBN1 gene has a 50% chance of inheriting Marfan syndrome. Both males and females are equally likely to inherit and be affected by autosomal dominant conditions.
However, there are rare case reports describing autosomal recessive fibrillin 1 gene (FBN1) mutations 22. In autosomal recessive pattern of genetic inheritance, Marfan syndrome only appears only in individuals who have inherited 2 copies of a mutated or altered FBN1 gene, one from each parent, located on a non-sex (autosomal) chromosome. Both parents of an affected individual are typically “carriers,” possessing one mutated copy and one working copy of the FBN1 gene, but do not themselves show symptoms of Marfan syndrome. In autosomal recessive inheritance if a child inherits one mutated FBN1 gene and one working copy, the child will be a carrier but will not have Marfan syndrome.
About 25% of patients with Marfan syndrome are the first in their family to have it, when this happens it is called a spontaneous mutation or “de novo mutation” 23, 24. These spontaneous mutation or “de novo mutation” Marfan syndrome cases are more severely affected. A de novo mutation is a genetic alteration that appears for the first time in an individual and is not inherited from either parent. These mutations can occur in a parent’s egg or sperm cell, or in the fertilized egg itself during early development, leading to a new genetic change in that person. De novo mutations are responsible for some genetic disorders and diseases, as they represent fresh genetic changes that were not present in the prior generations of a family.
- 75% of Marfan syndrome is most often inherited in an autosomal dominant pattern from a parent, who will have a 50% chance of passing the condition on to their children.
- 25% Marfan syndrome is due to a new random mutation (de novo mutation) in the FBN1 gene. These cases occur in people with no history of the disorder in their family.
- There are rare case reports describing autosomal recessive fibrillin 1 gene (FBN1) mutations 22. In autosomal recessive pattern of genetic inheritance, Marfan syndrome only appears only in individuals who have inherited 2 copies of a mutated or altered FBN1 gene, one from each parent, located on a non-sex (autosomal) chromosome. Both parents of an affected individual are typically “carriers,” possessing one mutated copy and one working copy of the FBN1 gene, but do not themselves show symptoms of Marfan syndrome. In autosomal recessive inheritance if a child inherits one mutated FBN1 gene and one working copy, the child will be a carrier but will not have Marfan syndrome.
Marfan syndrome affects men and women equally and occurs among all races and ethnic groups. Because it’s a genetic condition, the greatest risk factor for Marfan syndrome is having a parent with the disorder.
Education in the form of genetic counseling is particularly important for families with Marfan syndrome as there is a 50% chance of one affected parent passing the disease on to their child. If you or a family member are diagnosed with Marfan’s syndrome you will likely be referred to a genetic counselor or clinical geneticist for information regarding your disorder and screening programs. All close family members may be offered testing for the gene involved and be screened on a yearly basis for the development of clinical features.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 8. Marfan syndrome autosomal dominant inheritance pattern (75 percent of Marfan syndrome cases)
Marfan syndrome eyes
Most people with Marfan syndrome suffer from nearsightedness or myopia, and abnormal curvature of the eye or astigmatism. These can be notably high since the connective tissue defect can affect the cornea, lens, and growth of the eye. Other signs and symptoms include ectopia lentis, corneal thinning, flattened corneal curvature, cataracts, glaucoma, strabismus, and retinal detachment.
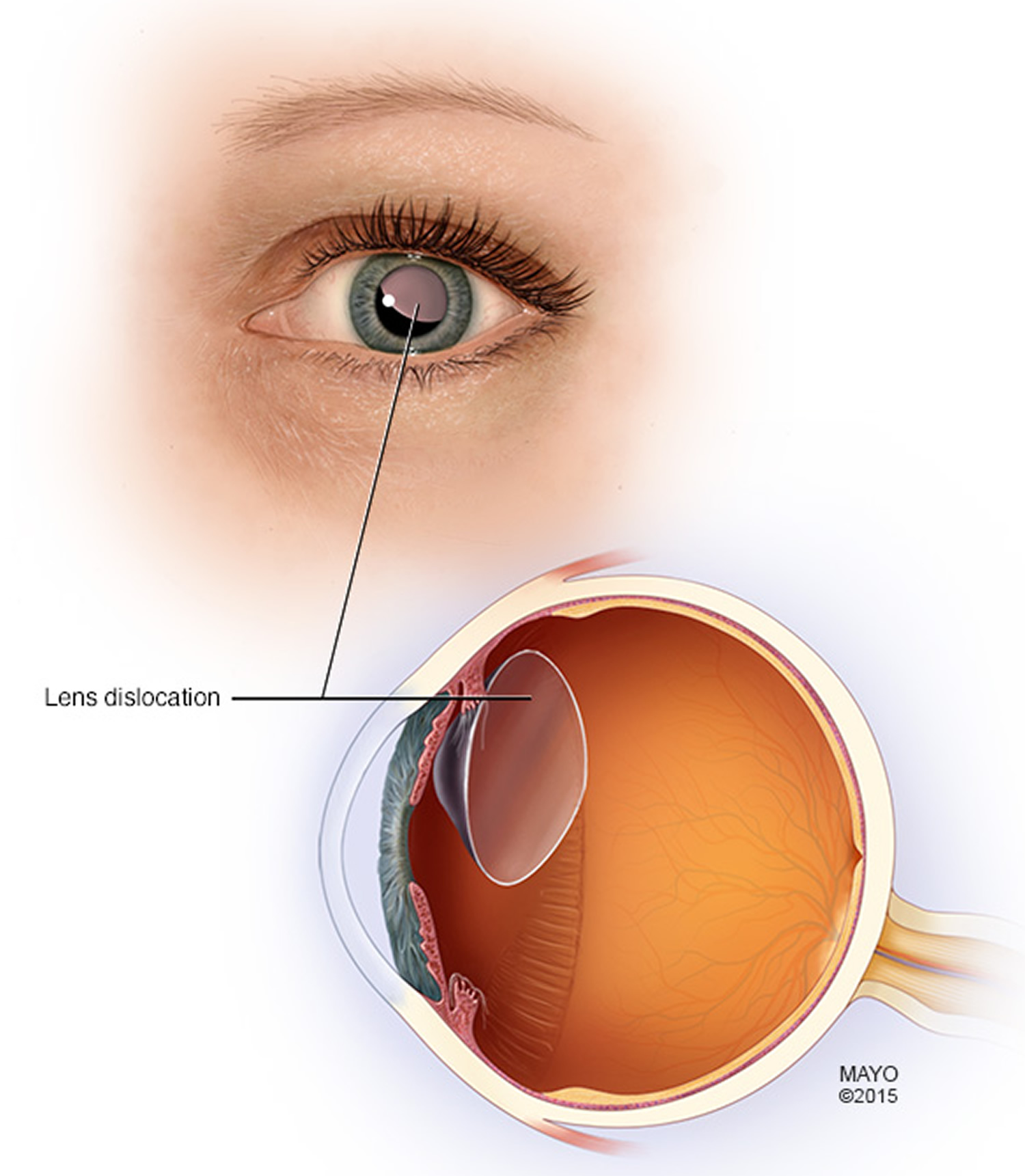
What is ectopia lentis?
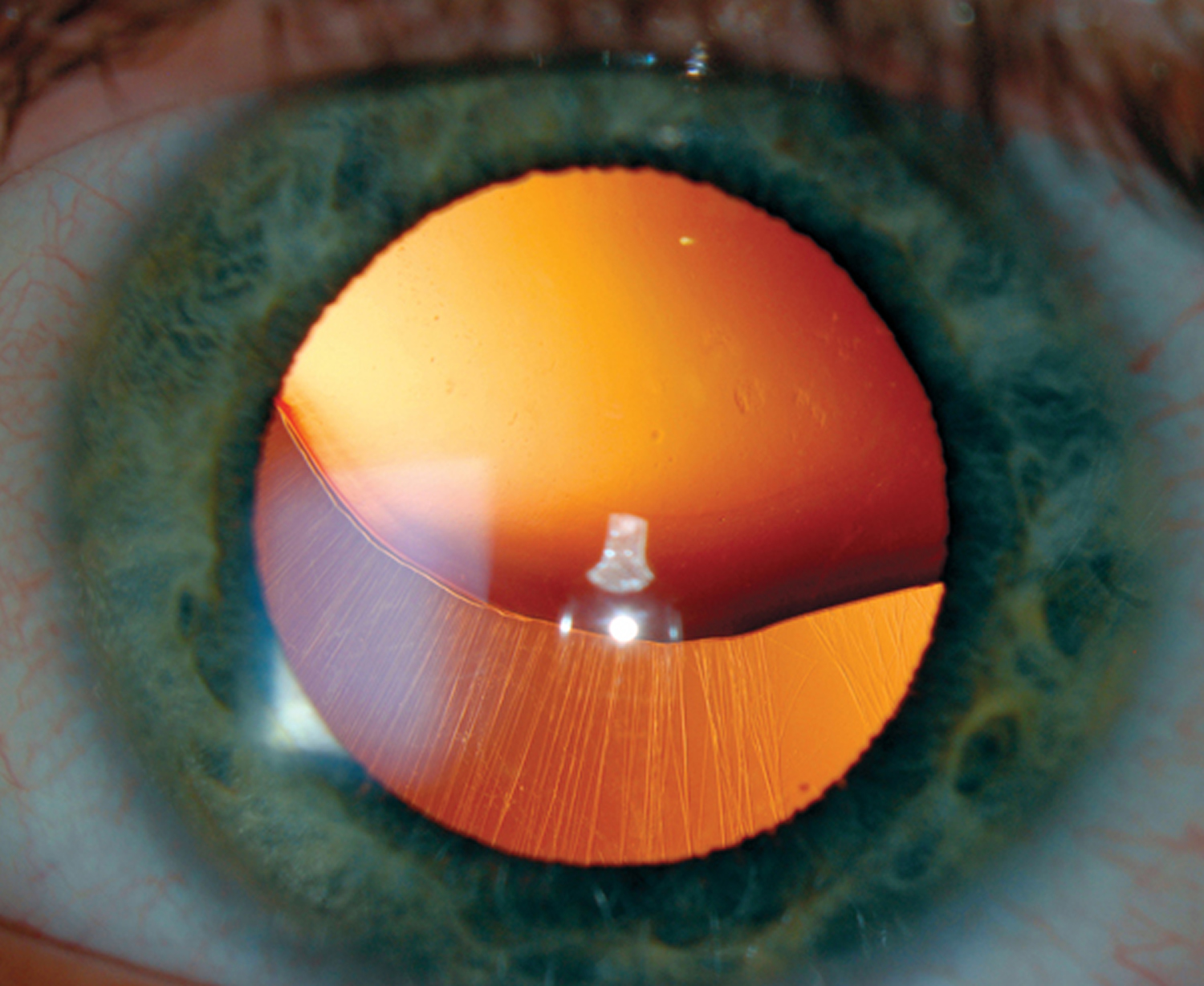
Figure 9. Marfan syndrome – Ectopia lentis (dislocated lens)
Marfan syndrome signs and symptoms
The signs and symptoms of Marfan syndrome vary greatly, even among members of the same family, because Marfan syndrome can potentially affect many systems of the body including the heart, blood vessels, skeleton, eyes, lungs and skin. Some people experience only mild effects, but others develop life-threatening complications, about 1 in 10 – experience more severe symptoms. In most cases, Marfan syndrome tends to worsen with age.
In some infants, Marfan’s syndrome may cause severe, rapidly progressive complications during infancy, often quickly affecting multiple organ systems early in life.
Marfan syndrome features may include:
- Tall and slender build
- Disproportionately long arms, legs and fingers
- A breastbone that protrudes outward or dips inward
- A high, arched palate and crowded teeth
- Heart murmurs
- Extreme nearsightedness
- An abnormally curved spine (scoliosis)
- Flat feet (pes planus)
Other features include hernias (part of the intestine protruding through a weakened part of the abdominal wall), and lung complications (e.g., sudden lung collapse, emphysema, asthma, sleep apnea).
Individuals with Marfan syndrome often develop distinct physical findings including an abnormally thin physique and disproportionately long, slender arms and legs (dolichostenomelia) due to overgrowth of the long bones. In addition, affected individuals usually have abnormally long, slender fingers (arachnodactyly). People with Marfan syndrome are usually very tall and thin in comparison to unaffected family members but not necessarily in comparison to the general population. Individuals with Marfan syndrome can lack muscle tone (hypotonia) and have little fat under the skin (subcutaneous fat).
A variety of bone malformations affect individuals with Marfan syndrome including overgrowth of the ribs, which can push the breastbone (sternum) inward resulting in a sunken chest (pectus excavatum) or outward resulting in a protruding chest (pectus carinatum). Additional symptoms include abnormally loose or flexible joints (joint hypermobility), flat feet (pes planus), fingers that are permanently bent or “fixed” and cannot extend or straighten fully (camptodactyly or clinodactyly), and reduced extension of the elbow. In some people, the joints may be unaffected or may become tight and stiff (contractures). Some individuals have an abnormally deep hip socket (acetabulum) with deep insertion of the head of the long bone (femur) of the leg (protrusio acetabulae) and signs of bone erosion. Many individuals with Marfan syndrome develop spinal abnormalities such as progressive curving of the spine (scoliosis) that may be mild or severe. Scoliosis may be associated with back pain in some affected individuals. In children, skeletal abnormalities may progress rapidly during phases of rapid growth, such as adolescence.
Individuals with Marfan syndrome may have several distinct facial features including a long, narrow skull (dolichocephaly), deep-set eyes (enophthalmos), an abnormally small jaw (micrognathia) that may be recessed farther back than normal (retrognathia), abnormally flat cheek bones (malar hypoplasia) and a downward slant to the eyes (downward slanting palpebral fissures). Affected individuals may also exhibit a highly arched roof of the mouth (palate), teeth that are crowded together and upper and lower teeth that do not meet (align) properly when biting (malocclusion).
Individuals with Marfan syndrome may have significant heart and blood vessels problems such as a common heart defect known as mitral valve prolapse. The mitral valve has two leaflets (or flaps) and is located between the left upper and left lower chambers (left atrium and left ventricle, respectively) of the heart. Mitral valve prolapse occurs when one or both flaps (cusps) of the mitral valve bulge or collapse backward (prolapse) into the left upper chamber (atrium) of the heart during ventricular contraction. In some people, this may allow leakage or the backward flow of blood from the left lower chamber of the heart (ventricle) back into the left atrium (mitral valve regurgitation). Often no associated symptoms are apparent (asymptomatic). However, mitral valve prolapse can result in chest pain, abnormal heart rhythms (arrhythmias), or evidence of inadequate heart function such as congestive heart failure, most often in association with prolonged and severe mitral regurgitation.
Additional heart and blood vessels findings include widening (aneurysm) and degeneration of the main artery that carries blood away from the heart (aortic aneurysm), tearing (aortic dissection) of the aorta so that blood seeps between the inner and outer layers of the aortic wall and backward flow of blood from the aorta into the lower left chamber (ventricle) of the heart (aortic regurgitation). If severe and left untreated, these heart abnormalities associated with Marfan syndrome can cause life-threatening complications such as rupture of the aorta and congestive heart failure. Some individuals may develop widening of the main artery of the lungs (pulmonary artery dilatation). This typically does not cause any problems in people with Marfan syndrome.
Individuals with Marfan syndrome commonly develop abnormalities of the eyes, especially nearsightedness (myopia), which may develop early in childhood and become progressively worse. Approximately 60 percent of individuals develop displacement of the lenses of the eyes away from the center of the eye (ectopia lentis). Ectopia lentis may occur at birth or later in life and may remain stable or become progressively worse.
Additional issues affecting the eyes in Marfan syndrome include an abnormally flat cornea (the front portion of the eyes through which light passes), underdevelopment of the colored portion of the eye (hypoplastic iris) and detachment of the nerve-rich membrane (retina) lining the back of the eyes (retinal detachment). Some individuals with Marfan syndrome are at risk for the early development of clouding of the lenses of the eyes (cataracts) or increased pressure and/or associated changes in the eyes (glaucoma). If left untreated, eye abnormalities can result in vision loss.
Some individuals with Marfan syndrome may develop distended air pockets near the top of the lungs (apical pulmonary blebs) which can predispose individuals to a leak of air within the chest cavity and lung collapse that occurs for no readily apparent reason (spontaneous pneumothorax). In some people, pneumothorax can recur in the same lung or even the opposite lung (recurrent pneumothorax). Marfan syndrome can be associated with blockage of the upper airway during sleep (obstructive sleep apnea) that can impair lung and heart function if severe and left untreated.
Some affected individuals may develop widening or bulging of the sac (dura) that surrounds the spinal cord (dural ectasia). This condition usually does not cause symptoms (asymptomatic) but has been associated with lower back pain and can cause pinching of a nerve leading to abnormal sensations or muscle performance in the legs. Affected individuals may also developed stretch marks (striae atrophicae) of the skin without an obvious cause. Some affected individuals may have an inguinal, umbilical or surgical hernia, in which a weakened portion of the pelvic or abdominal wall shows external bulging, sometimes allowing protrusion of a small segment of the intestines.
Skeleton
Someone with Marfan syndrome may have several distinct physical characteristics. They may be:
- tall and slim, with long, thin arms and legs
- have loose and very flexible joints
Other physical characteristics of Marfan syndrome can include:
- a small lower jaw
- a high, arched palate (roof of the mouth)
- deep-set eyes
- flat feet
- a breastbone (sternum) that either protrudes outwards or indents inwards
- crowded teeth
Scoliosis
Marfan syndrome can cause the spine to become abnormally curved to the sides. This is known as scoliosis.
Curvature of the spine can cause long-term backache. In severe cases, it can also make breathing difficult as the spine may press against the heart and lungs.
Spondylolisthesis
Spondylolisthesis is where one of the bones in your spine (a vertebra) slips forward over another vertebra.
This usually occurs in the lower spine, and can cause back pain and stiffness. Anyone can develop spondylolisthesis, but it more commonly affects people with Marfan syndrome.
Dural ectasia
The dura is the membrane that lines your brain and spinal cord. Dural ectasia is a condition where the dura becomes weakened and expands outwards.
People with Marfan syndrome are at particular risk of developing dural ectasia. As the membrane expands, it can press on the vertebrae in your lower back, which can cause:
- backache
- headache
- numbness or pain in your legs
Eyes
Many people with Marfan syndrome have some type of vision problem.
Lens dislocation affects half of all people with the syndrome. This is where the eye’s lens, the transparent structure that sits behind the pupil and focuses light, falls into an abnormal position.
Other possible eye-related symptoms of Marfan syndrome include:
- myopia – short-sightedness
- glaucoma – increased pressure in the eyeball which, left untreated, can cause permanent vision loss
- cataracts – where cloudy patches develop in the eye’s lens, causing blurred or misty vision
- retinal detachment – where the light-sensitive layer of cells at the back of your eye (retina) begins to pull away from the blood vessels that supply it with oxygen and nutrients
Cardiovascular system
Marfan syndrome can affect the cardiovascular system, which is made up of your heart and blood vessels. It’s particularly serious if your aorta and heart valves are affected.
Aorta
The aorta is the main artery in the body. It runs from your heart, down the centre of your chest, and through your abdomen.
In people with Marfan syndrome, the walls of the aorta are weak. This can sometimes cause the aorta to enlarge and balloon, which is known as an aortic aneurysm.
In severe cases, the aorta can split (rupture), causing potentially fatal internal bleeding.
Heart valves
Your heart has four chambers that pump blood to and from the rest of the body. To control the flow of blood through your heart’s chambers, your heart has four valves:
- mitral valve
- aortic valve
- tricuspid valve
- pulmonary valve
These valves act as one-way gates, allowing blood to flow through in one direction. In some people with Marfan syndrome, the mitral or tricuspid valves don’t close properly and blood leaks back through the valve.
The aortic valve may also leak, leading to the main pumping chamber (left ventricle) gradually becoming enlarged.
Stretch marks
Stretch marks are pink, red, or white streaks in the skin. They can appear when you gain or lose weight quickly, when you have a growth spurt during childhood, or during pregnancy.
People with Marfan syndrome often develop stretch marks because the tissue in their skin is weakened and the skin isn’t as elastic as it should be.
If you have Marfan syndrome, stretch marks are most likely to appear on your:
- shoulders
- hips
- lower back
Over time, they’ll gradually fade to a silvery color and will be difficult to see.
If you suffer Marfan’s syndrome you may have noticed the following features:
- Delayed gross and motor development due to laxity of ligaments.
- You may have been diagnosed with a murmur previously.
- Abnormal heart rhythms.
- Sudden onset of thoracic (chest) pain due to aortic dissection.
- Lower back pain- Possibly with burning, numbness or weakness in the legs due to an abnormality of a membrane lining the spinal canal. This can also cause headaches and other neurologic deficits.
- Joint pain in adults.
- Shortness of breath, palpitations and substernal pain in severe pectus excavatum (deformity of the chest wall).
- Visual problems or loss of vision due to lens dislocation or retinal detachment.
Skeletal abnormalities are usually the most obvious feature of Marfan’s. Individuals are typically exceptionally tall, with long arms and legs, and elongated tapering fingers and toes. The ligaments in hand and foot joints are lax, and individuals may appear to be double jointed due to the increased joint mobility. A number of spinal abnormalities may be present, including kyphosis (increased outwards curvature of the spine) or scoliosis (sideways curvature of the spine). There maybe also be abnormalities of the chest wall, which may be depressed inwards (pectus excavatum) or outwards (pectus carinatum).
Eye changes may also be present. Most common is dislocation of the lens in both eyes (ectopia lentis). This is very uncommon, and if present should raise the suspicion of Marfan’s syndrome.
The most serious manifestation of Marfan’s syndrome are heart complications. Dilatation of the aorta (widening and structural weakening of the aorta) may occur, increasing the risk of an aortic dissection – a rupture through the aorta causing massive hemorrhage. The heart valves may also be affected. Deficient connective tissue support results in floppy valves, which may predispose to heart failure. An abnormal heart rhythm may be present.
Marfan syndrome complications
Because Marfan syndrome can affect almost any part of your body, it may cause a wide variety of complications.
Marfan syndrome complications include:
- Aortic aneurysm and dissection
- Mitral valve prolapse
- Mitral regurgitation
- Aortic regurgitation
- Lens subluxation (ectopia lentis)
- Cataract, glaucoma, and retinal detachment
- Spontaneous pneumothorax
- Inguinal hernias
- Scoliosis
Cardiovascular complications
The most dangerous complications of Marfan syndrome involve the heart and blood vessels. Faulty connective tissue can weaken the aorta — the large artery that arises from the heart and supplies blood to the body.
- Aortic aneurysm. The pressure of blood leaving your heart can cause the wall of your aorta to bulge out, like a weak spot in a tire. In people who have Marfan syndrome, this is most likely to happen at the aortic root — where the artery leaves your heart.
- Aortic dissection. The wall of the aorta is made up of layers. Dissection occurs when a small tear in the innermost layer of the aorta’s wall allows blood to squeeze in between the inner and outer layers of the wall. This can cause severe pain in the chest or back. An aortic dissection weakens the vessel’s structure and can result in a rupture, which may be fatal.
- Valve malformations. People who have Marfan syndrome also are more likely to have problems with their heart valves, which may be malformed or overly elastic. When heart valves don’t work properly, your heart often has to work harder to compensate. This can eventually lead to heart failure.
Eye complications
Eye complications may include:
- Lens dislocation. The focusing lens within your eye can move out of place if its supporting structures weaken. The medical term for this problem is ectopia lentis, and it occurs in more than half the people who have Marfan syndrome.
- Retinal problems. Marfan syndrome also increases the risk of a detachment or tear in the retina, the light-sensitive tissue that lines the back wall of your eye.
- Early-onset glaucoma or cataracts. People who have Marfan syndrome tend to develop these eye problems at a younger age. Glaucoma causes the pressure within the eye to increase, which can damage the optic nerve. Cataracts are cloudy areas in the eye’s normally clear lens.
Skeletal complications
Marfan syndrome increases the risk of abnormal curves in the spine, such as scoliosis. It can also interfere with the normal development of the ribs, which can cause the breastbone to either protrude or appear sunken into the chest. Foot pain and low back pain are common with Marfan syndrome.
Complications of pregnancy
Marfan syndrome can weaken the walls of the aorta, the main artery that leaves the heart. During pregnancy, a woman’s heart is pumping more blood than usual, and this can put extra stress on a woman’s aorta — which increases the risk of a deadly dissection or rupture.
Marfan syndrome diagnosis
Marfan syndrome can be challenging for doctors to diagnose because many connective tissue disorders have similar signs and symptoms. Even among members of the same family, the signs and symptoms of Marfan syndrome vary widely — both in their features and in their severity.
Certain combinations of symptoms and family history must be present to confirm a diagnosis of Marfan syndrome. In some cases, a person may have some features of Marfan syndrome, but not enough of them to be diagnosed with the disorder.
No universal, specific diagnostic test exists for Marfan syndrome despite the identification of the causative gene. A diagnosis is made based upon a detailed patient and family history, a thorough clinical evaluation, and a variety of specialized tests performed to identify key findings associated with Marfan syndrome. Different criteria have been proposed for classifying someone as having Marfan syndrome. The most recent published criteria, the Ghent criteria, were published in 2010. According to these guidelines, the presence of aortic root aneurysm, eye lens dislocation, or a family history of definite Marfan syndrome weigh heavily in the diagnosis of Marfan syndrome, with an additional potential contribution of other findings throughout the body. Molecular genetic testing looking for a variant in the FBN1 gene can aid in the diagnosis of Marfan syndrome, but identifying a variant is not sufficient to establish the diagnosis in the absence of sufficient physical findings or family history.
Individuals suspected of having Marfan syndrome will usually undergo a complete skeletal examination, a heart examination including a test that uses sound waves to produce images of the heart (echocardiogram) and a specialized examination of the eyes (slit-lamp eye examination). A slit-lamp allows an eye doctor to examine the eyes under high magnification to detect lens dislocation and other eye issues. It is essential that this comprehensive diagnostic evaluation be coordinated by someone very familiar with Marfan syndrome and related diagnoses.
Ghent criteria (Ghent nosology)
Your doctor may compare the signs and symptoms against the Ghent criteria also called Ghent nosology 34. This is a diagnostic tool that helps doctors and other healthcare professionals tell the difference between Marfan syndrome and other similar syndromes.
The Ghent criteria (Ghent nosology) consists of major and minor criteria. The major criteria are features or symptoms common in people with Marfan syndrome that are rare in people who don’t have it.
Minor criteria are features or symptoms present in people with Marfan syndrome, but are also present in people who don’t have it.
To be diagnosed with Marfan syndrome using the Ghent criteria, you must have a number of different symptoms.
- If you have a family history of Marfan syndrome, you’ll need to have one of the major criteria and one of the minor criteria.
- If you don’t have a family history of Marfan syndrome, you’ll need to have two major criteria and one of the minor criteria.
Some of the major and minor criteria used to help diagnose Marfan syndrome are listed below.
Major criteria
Major criteria can include:
- an enlarged aorta
- a tear in the aorta
- dislocation of the lens of the eye
- a family history of the syndrome
- at least four skeletal problems, such as flat feet or curved spine (scoliosis)
- enlargement of the lining that surrounds part of the spinal cord (dural ectasia)
Minor criteria
Minor criteria can include:
- near-sightedness (myopia)
- unexplained stretch marks
- loose joints
- a long, thin face
- a high, arched palate (roof of the mouth)
Ghent-2 criteria (revised Ghent nosology)
In 2010 the Ghent criteria (Ghent nosology) underwent revision due to various limitations, including insufficient validation, limited applicability to children, and an inability to exclude syndromes such as Loeys-Dietz syndrome and Shprintzen-Goldberg syndrome 35. The 2010 revised Ghent nosology (Ghent-2 criteria) puts greater weight on aortic root dilatation/dissection and ectopia lentis as the cardinal clinical features of Marfan syndrome and testing for mutations in FBN1 gene 35.
The revised Ghent nosology (Ghent-2 criteria) includes the following scoring system for systemic features 35:
- Wrist and thumb sign: 3 points
- Wrist or thumb sign: 1 point
- Pectus carinatum deformity: 2 points
- Pectus excavatum or chest asymmetry: 1 point
- Hindfoot deformity: 2 points
- Plain pes planus: 1 point
- Pneumothorax: 2 points
- Dural ectasia: 2 points
- Protrusio acetabuli: 2 points
- Reduced upper segment/lower segment ratio and increased arm span/height and no severe scoliosis: 1 point
- Scoliosis or thoracolumbar kyphosis: 1 point
- Reduced elbow extension (equal to 170 degrees with full extension): 1 point
- At a minimum, 3 of the following 5 features: Dolichocephaly (reduced cephalic index or head width/length ratio), enophthalmos, down-slanting palpebral fissures, malar hypoplasia, retrognathia): 1 point
- Skin striae: 1 point
- Myopia greater than 3 diopters: 1 point
- Mitral valve prolapse: 1 point
A systemic score equal to 7 indicates significant systemic involvement.
In patients with no family history of Marfan syndrome, the presence of 1 of any of the following criteria is diagnostic for Marfan syndrome 35:
- Aortic criterion (aortic root dissection or diameter Z equal to 2) and ectopia lentis*
- Aortic criterion (aortic root dissection or diameter Z equal to 2) and a causal FBN1 mutation
- Aortic criterion (aortic diameter Z equal to 2 or aortic root dissection) and a systemic score equal to 7*
- Ectopia lentis and an identified causal FBN1 mutation in an individual with an aortic aneurysm
In patients with a family history of Marfan syndrome, the presence of 1 of any of the following criteria is diagnostic for Marfan syndrome 35:
- Ectopia lentis
- A systemic score equal to 7 points*
- Aortic criterion (aortic diameter Z equal to 3 below 20 years old, Z equal to 2 above 20 years, or aortic root dissection)*
For criteria with an asterisk (*), the diagnosis of Marfan syndrome can be made only in the absence of discriminating features of Shprintzen-Goldberg syndrome, Loeys-Dietz syndrome, or vascular Ehlers-Danlos syndrome (vEDS) and after TGFBR1/TGFBR2, collagen biochemistry, or COL3A1 testing if indicated. Later data suggest that additional gene mutations, including those in SMAD3, TGFB2, and SKI, should also be excluded.
The revised Ghent nosology recommends the following categories for individuals younger than 20 years old with features of Marfan syndrome who do not meet diagnostic criteria for Marfan syndrome 35:
- Nonspecific connective tissue disorder applies if the systemic score is less than 7 or aortic root measurements are borderline (Z less than 3) without an FBN1 mutation.
- Potential Marfan syndrome applies if an FBN1 mutation is identified in a sporadic or familial case but the aortic root Z-score is less than 3.
Marfan syndrome test
Heart tests
If your doctor suspects Marfan syndrome, one of the first tests he or she may recommend is an echocardiogram. This test uses sound waves to capture real-time images of your heart in motion. It checks the condition of your heart valves and the size of your aorta. Other heart-imaging options include computerized tomography (CT) scans and magnetic resonance imaging (MRI).
If you are diagnosed with Marfan syndrome, you’ll need to have regular echocardiograms to monitor the size and condition of your aorta.
Eye tests
Eye exams that may be needed include:
- Slit-lamp exam. This test checks for lens dislocation, cataracts or a detached retina. Your eyes will need to be completely dilated with drops for this exam.
- Eye pressure test. To check for glaucoma, your eye doctor may measure the pressure inside your eyeball by touching it with a special tool. Numbing eyedrops are usually used before this test.
Genetic testing
If findings from standard exams for Marfan syndrome aren’t clear-cut, genetic testing can be helpful.
Although the gene that causes Marfan syndrome has been identified, it can change (mutate) in more than 3,000 different ways. Genes are single units of genetic material.
You may also want to talk to a genetic counselor before starting a family, to see what your chances are of passing on Marfan syndrome to your future children.
A genetic test can be used to examine the gene responsible for Marfan syndrome. It’s able to detect an error that causes the syndrome in 99% of those affected. However, the test is expensive and takes three months to complete.
In most cases, a diagnosis of Marfan syndrome will be made from the physical features and symptoms of the syndrome.
Prenatal testing
Once a gene mutation for Marfan syndrome has been found, and you’re going to become a parent, you may want to have your unborn baby tested to find out whether they also have the syndrome.
There’s a one in two (50%) chance of the baby inheriting the syndrome.
To do this, two possible tests can be used – chorionic villus sampling (CVS) or amniocentesis.
Chorionic villus sampling
Prenatal testing for Marfan syndrome can be carried out approximately 10 to 12 weeks into the pregnancy using chorionic villus sampling (CVS).
Chorionic villus sampling involves taking a small sample of cells from the organ that links the mother’s blood supply with her unborn baby’s (the placenta) through the entrance of the womb. The sample can then be tested for genetic conditions.
Amniocentesis
Amniocentesis can also be used to test for Marfan syndrome. The test is carried out about 16 to 18 weeks into the pregnancy and involves taking a small sample of amniotic fluid for examination. Amniotic fluid surrounds the unborn baby in the womb.
Caution
Although prenatal tests may show whether your child has the defective gene that causes Marfan syndrome, the tests won’t give any indication as to how serious their symptoms will be. Generally, the baby will be affected to the same extent as other people in their family.
The severity of Marfan syndrome in the parent is an indication of how severe it will be in the child. Your child may only experience very mild symptoms, despite having the genetic mutation. This is because the expression of the gene can vary, even within the same family.
In some cases, the results of chorionic villus sampling or amniocentesis could be negative, suggesting that your child doesn’t have the defective gene. However, your child may have a different genetic mutation that wasn’t tested for, but could still cause Marfan syndrome.
Preimplantation genetic diagnosis
Preimplantation genetic diagnosis is a technique where eggs and sperm are harvested from the parents so embryos can be created in a laboratory.
Only unaffected embryos are available for implantation in the womb. The process takes about 12 months.
Preimplantation genetic diagnosis is only an option after a person with Marfan syndrome has been identified as having a Marfan gene mutation and wants to become a parent.
Marfan syndrome differential diagnosis
Symptoms of the following disorders can be similar to those of Marfan syndrome. Comparisons are essential to arrive at a correct diagnosis.
Loeys-Dietz syndrome is a rare inherited connective tissue disorder characterized by a variety of symptoms that overlap with Marfan syndrome. Individuals with Loeys-Dietz syndrome have skeletal and cardiovascular abnormalities. Affected individuals may experience bulging of the wall of the aorta (aneurysm), tearing (dissection) of the aorta or rupture of the aorta. Unlike Marfan syndrome, the aorta in individuals with Loeys-Dietz syndrome is prone to tearing or rupturing early in childhood and at a relatively small size. In Loeys-Dietz syndrome the blood vessels typically follow a very winding course (tortuosity), and aneurysms and tears of blood vessels can occur throughout the entire arterial tree. Loeys-Dietz syndrome is inherited in an autosomal dominant pattern and is caused by variants in at least seven genes that are all known to influence the activity of transforming growth factor beta (TGF-β) (TGFBR1, TGFBR2, SMAD3, SMAD2, TGFB2, TGFB3 and IPO8). While all patients with variants in these genes do not have outward discriminating features of Loeys-Dietz syndrome, they should have imaging studies to look for vascular issues throughout the arterial tree from head through pelvis and are at risk to have children with more typical Loeys-Dietz syndrome features.
Shprintzen-Goldberg syndrome shares many features with Marfan syndrome and Loeys-Dietz syndrome including overgrowth of the long bones (dolichostenolelia), long and slender fingers (arachnodactyly), abnormal curvature of the spine (scoliosis), a sunken or protruding chest (pectus excavatum or pectus carninatum), widening of the spinal sac (dural ectasia), loose joints, wide spacing of the eyes (hypertelorism), premature fusion of the sutures of the skull (craniosynostosis) and a highly arched palate. While aortic root enlargement (aneurysm) can be seen in Shprintzen-Goldberg syndrome, it is less frequent and typically less severe, when compared to Marfan syndrome or Loeys-Dietz syndrome. Unlike either Marfan syndrome or Loeys-Dietz syndrome, most people with Shprintzen-Goldberg syndrome have at least some degree of developmental delay or intellectual disability. Shprintzen-Goldberg syndrome is caused by variants in the SKI gene that influences the activity of transforming growth factor beta (TGF-β).
Beals syndrome also known as congenital contractural arachnodactyly, is an extremely rare inherited connective tissue characterized by fixed flexion (contracture) of certain joints (e.g., fingers, elbows, knees, and hips); abnormally long, slender fingers and toes (arachnodactyly); and/or abnormally shaped ears resulting in a “crumpled” appearance. Contractures of joints tend to improve with age in Beals syndrome. In addition, affected individuals may exhibit front-to-back and side-to-side curvature of the spine (kyphoscoliosis); feet that are abnormally positioned (talipes equinovarus or clubfoot); outward displacement of the fingers (ulnar deviation of the fingers); and an abnormally short neck. There are extremely rare reports of displacement of the lens of the eye (ectopia lentis) or severe heart abnormalities in patients with Beals syndrome; the exceptional nature of these cases warrants further study to determine if these findings are truly related to the diagnosis of Beals syndrome. Some affected individuals may have a deformity of the valve on the left side of the heart (mitral valve prolapse). Beals syndrome is inherited in an autosomal dominant pattern and is caused by variants in the FBN2 gene.
Ehlers-Danlos syndrome (EDS) is a group of inherited connective tissue disorders characterized by defects of the major structural protein in the body (collagen). Collagen, a strong and rigid protein, plays an essential role in holding together and strengthening the tissues of the body. Due to defects of collagen, primary Ehlers-Danlos syndrome (EDS) findings include abnormally flexible, loose joints (articular hypermobility) that may easily become dislocated and unusually loose, thin, stretchy (elastic) and/or fragile skin. Some forms of Ehlers-Danlos syndrome (EDS) can include weakness of tiny blood vessels (capillaries) and other tissues of the body, leading to easy bruising, hernias and other findings.
Mitral valve prolapse (MVP) is a common heart condition in which one or both of the flaps (cusps) of the mitral valve bulge or collapse backward (prolapse) into the left atrium during ventricular contraction. In some people, this may allow leakage or the backward flow of blood from the left ventricle back into the left atrium (mitral regurgitation). The exact underlying mechanism responsible for mitral valve prolapse remains unknown. In many affected individuals, mitral valve prolapse appears to occur in the absence of an associated disorder or syndrome (condition that involves multiple parts of the body). Sometimes mitral valve prolapse occurs in families in association with subtle connective tissue findings in other parts of the body (mitral valve prolapse syndrome). Evidence indicates that both the isolated and syndromic forms of mitral valve prolapse are sometimes familial, suggesting autosomal dominant inheritance. Mitral valve prolapse can also occur in association with other defined connective tissue disorders including Marfan syndrome, Loeys-Dietz syndrome, Shprintzen-Goldberg syndrome and other conditions. In many individuals with mitral valve prolapse, no associated symptoms are apparent (isolated mitral valve prolapse). However, in other people, the condition may result in chest pain, abnormal heart rhythms (arrhythmias) and signs of congestive heart failure, especially when valve leakage is longstanding (chronic) and severe. Mitral valve prolapse is often associated with a characteristic click and/or a whooshing sound (murmur) that may be detected through use of a stethoscope during physical examination. If leakage through the mitral valve (mitral regurgitation) is severe, the extra work that the heart has to perform can lead to poor performance of the heart muscle (heart failure).
Homocystinuria is a rare inherited metabolic disorder characterized by an excess of the compound homocystine in the blood and urine. Homocystinuria may result from deficiency of any of several enzymes involved in the conversion of the essential amino acid methionine to another amino acid called cysteine or, less commonly, impaired conversion of the compound homocysteine to methionine. Enzymes are proteins that accelerate the rate of chemical reactions in the body. Certain amino acids, which are the chemical building blocks of proteins, are essential for proper growth and development. In most cases, homocystinuria is caused by reduced activity of an enzyme known as cystathionine beta-synthase (CBS). Due to deficiency of the cystathionine beta-synthase (CBS) enzyme, infants with homocystinuria may fail to grow and gain weight at the expected rate (failure to thrive) and have developmental delay. By approximately age three, additional, more specific findings may become apparent. These may include dislocation (subluxation) of the lens of the eyes (ectopia lentis), associated “quivering” (iridodonesis) of the colored region of the eyes (iris), severe nearsightedness (myopia), and other eye (ocular) abnormalities.
Familial aortic aneurysm also known as Familial Thoracic Aortic Aneurysm and Dissection (FTAAD) is a rare inherited connective tissue disorder where a person’s main artery (the aorta) develops a bulge (aneurysm) or a tear (dissection) due to genetic changes. These are serious, potentially life-threatening conditions where the weakened aorta can rupture, causing massive bleeding and internal injury. Some affected individuals can show subtle cysts of the colored portion of the eye (iris floculi) or a visible capillary network below the skin with a honeycomb appearance (livido reticularis). A subset of people with Familial Thoracic Aortic Aneurysm and Dissection (FTAAD) can have an aortic valve that forms with only two cusps (bicuspid aortic valve) instead of the normal three cusps (tricuspid aortic valve). The base of the aorta (aortic root) is the most common site of enlargement (aneurysm), but other aortic segments and even blood vessels outside of the chest can more rarely show involvement. Mutations in any of several genes are associated with familial Familial Thoracic Aortic Aneurysm and Dissection (FTAAD). Mutations in the ACTA2 gene have been identified in 14 to 20 percent of people with Familial Thoracic Aortic Aneurysm and Dissection (FTAAD), and TGFBR2 gene mutations have been found in 2.5 percent of affected individuals. Mutations in several other genes account for smaller percentages of cases and only a few have been identified. Familial Thoracic Aortic Aneurysm and Dissection (FTAAD) most often follows an autosomal-dominant inheritance pattern, meaning a 50% risk of passing it to each child, and can also be associated with other connective tissue disorders. The age at which problems arise tends to be later and more variable than in Marfan syndrome or Loeys-Dietz syndrome, and some people who inherit a gene variant might never show a vascular problem. Aortic root aneurysms tend to tear or rupture at a size similar to that in Marfan syndrome, and many of the same management principles apply.
Marfan syndrome treatment
As Marfan syndrome affects several different parts of the body, your treatment programme will involve a number of healthcare professionals, who may include:
- a geneticist – a specialist in genetic disorders
- a genetic counselor – who provides information, emotional support and guidance to people who’ve been diagnosed with a genetic condition
- a cardiologist – a specialist in heart conditions
- an ophthalmologist – a specialist in conditions that affect the eyes
- an orthopedic surgeon – a surgeon who specialises in treating conditions that affect the muscles, joints and bones
- a pediatrician – a specialist in treating babies and children up to the age of 16
You’ll usually be assigned a doctor to co-ordinate your treatment programme and ensure every aspect of the syndrome is closely monitored and, if necessary, treated.
Individuals with Marfan syndrome are advised to avoid strenuous activities (heavy lifting and any exercise that increases the strain on the aorta produced by rapid or vigorous beating of the heart or increased blood pressure) and contact sports due to risk of heart complications. Highly competitive sports for those involving isometric work such as weightlifting, climbing steep inclines, gymnastics, and pull-ups should be avoided due to risks of increased blood pressure and sudden death. Restriction of such activities can slow the rate of the widening of the aorta (aortic dilatation) and decrease the tendency for aortic tear (dissection). Non-strenuous activities and sports such as golf, walking and fishing are recommended. In general, moving types of exercises performed in moderation are thought to be good for people with Marfan syndrome. Such exercises, performed regularly, will naturally lower heart rate and blood pressure.
It may also be necessary to change your job to a more sedentary one as you may find yourself easily fatigued or the visual and painful skeletal changes may limit your work.
Patients with Marfan’s syndrome are usually given medications long-term to help lower the risk of heart complications associated with Marfan syndrome. The main class of drugs used are called beta-blockers (beta-adrenergic receptor blocking drugs) which reduce the dilation of the aortic root. Such drugs help to reduce the strength and frequency of the contractions of the heart and lower blood pressure. In doing so, they may reduce the strain on the walls of the aorta. Beta-blockers may delay the need for heart surgery. The dosage needs to be adjusted to the individual patient’s needs, and therapy should be closely monitored. Some individuals may not be able to tolerate beta-blockers due to their low blood pressure (hypotension) or they have other medical conditions such asthma or depression may not be able to take them (contraindicated). Losartan (Cozaar) or irbesartan, a newer blood pressure medication, can be prescribed for protecting your aorta.
Every person with Marfan’s syndrome should have long term monitoring of heart complications that include regular blood pressure checks and at least a yearly echocardiogram to check the size and function of the heart and aorta. Your cardiologist will determine the frequency of these visits that is suitable for you depending on the severity of your condition. However, patients are usually followed up every six months to one year.
Surgical repair of the aorta may eventually become necessary if the aorta has severely widened or developed a tear (dissection). Preventive (prophylactic) surgery is recommended when the diameter of the aorta reaches 5 centimeters in older children or adults, when the rate of widening reaches 1 centimeter a year, or when there is severe or progressive backflow (regurgitation) of blood through the aortic valve. Surgery may also be necessary for leakage of the mitral valve. Replacement of the aortic valve with an artificial valve may be performed (composite graft repair); however, this surgery requires the lifelong use of medications to prevent blood clots (anticoagulation). In recent years, some physicians have preferred to use valve-sparing surgery (i.e., reimplantation of the natural aortic valve within a Dacron tube used to replace the enlarged segment of the aorta). Studies are underway to assess the durability of valve-sparing procedures, but early-to-intermediate data are encouraging.
Surgery to repair or replace the mitral valve in individuals who experience severe mitral valve regurgitation may become necessary. Cardiovascular problems related to Marfan syndrome increase susceptibility to repeated bacterial infections such as infections of the heart valves (bacterial endocarditis). Leaking heart valves are more prone to infection with bacteria. While it had been common practice to treat patients with leaking heart valves with antibiotics before dental work or other procedures expected to contaminate the blood stream with bacteria, the American Heart Association recently withdrew this recommendation for most people. Given the predisposition of people with Marfan syndrome and other connective tissue disorders to progressive leakage through multiple heart valves, many physicians who routinely care for such individuals continue to recommend that antibiotics be used before dental work or other procedures expected to introduce bacteria into the bloodstream.
Skeletal abnormalities such as scoliosis and deformity of the chest may represent serious problems for people with Marfan syndrome. Braces may be tried to correct skeletal curving (scoliosis) in some people but can be ineffective. Individuals with curvature of the spine of more than Cobb angle of 10 degrees should be followed by an orthopedic surgeon. Surgical stabilization of the spine may be needed if the curvature is severe or progressive. A sunken chest (pectus excavatum) may be surgically corrected for cosmetic reasons or, in very rare severe cases, to avoid medical complications.
The eyes require careful attention with yearly ophthalmologic exams from early childhood. Failure to detect any of the several abnormalities that can affect the eyes may result in poor vision and other visual impairment. Increased risk of retinal detachment does demand special attention. The eyes should receive special protection from injury during work or sports. Sports that may involve trauma to the head, such as football, boxing and diving, should be avoided. Displacement of the lenses may be treated with eyeglasses or contact lenses. Some individuals such as those with a completely loose lens or with a displaced lens that disrupts vision may require surgical intervention. A detached retina can sometimes be corrected, especially if detected early.
Genetic counseling is recommended for affected individuals and their families. Other treatment is symptomatic and supportive.
In the past, people who had Marfan syndrome rarely lived past 40. With regular monitoring and modern treatment, most people with Marfan syndrome can now expect to live a more normal life span.
Children with Marfan syndrome Management and Treatment
All children with Marfan syndrome or possible case of Marfan syndrome (children often have an evolving signs and symptoms and commonly need to be followed for several years before a Marfan syndrome diagnosis can be confirmed) should be regularly assessed by echocardiography, optometry, and skeletal survey as the child grows 36. Early referral to a cardiologist and clinical geneticist is key. All children with Marfan syndrome should undergo a yearly evaluation with a pediatric cardiologist to include physical exam and imaging of the aorta 37. The preferred imaging modality is echocardiography in the majority of patients. In patients who are unable to be properly imaged with echocardiography due to either obesity or extreme pectus deformity, MRI or CT scanning can be used yearly for follow up; though MRI is preferred to avoid radiation exposure whenever practical 37. In patients with an aortic root dimension above 4 cm, aortic root growth of more than 0.5 cm/year, or those involved in competitive sports, it is recommended to obtain aortic imaging every 6 months 37.
Lifestyle modifications are another mainstay in the management of Marfan syndrome. It is generally advised that children with Marfan syndrome avoid isometric exercise, high impact sports, and competitive sports due to the risk of aortic dissection, long bone fractures, and retinal detachment. Isometric exercises involve contracting a muscle or muscle group to create tension without changing the length of the muscle or the angle of the joint. Examples include a leg lift, plank, wall sit, glute bridge or dead hang. During isometric exercises, the muscle doesn’t noticeably change length. The affected joint also doesn’t move. However, most children should be encouraged to remain active with aerobic activities performed in moderation 38.
The commonly prescribed medications for Marfan syndrome patients are beta-blockers (beta-adrenergic receptor blocking drugs) which reduce the dilation of the aortic root 39. Such drugs help to reduce the strength and frequency of the contractions of the heart and lower blood pressure. In doing so, they may reduce the strain on the walls of the aorta. Beta-blockers may delay the need for heart surgery. The dosage needs to be adjusted to the individual patient’s needs, and therapy should be closely monitored. Some individuals may not be able to tolerate beta-blockers due to their low blood pressure (hypotension) or they have other medical conditions such asthma or depression may not be able to take them (contraindicated).
A second class of blood pressure medication called angiotensin receptor blockers (ARBs) is commonly used in the treatment of cardiovascular problems associated with Marfan syndrome 31. This includes medications such as losartan or irbesatan can be prescribed for protecting your aorta.
If the decision is made to use beta-blockers it must be titrated to clinical guidelines. While studies using animal models of Marfan syndrome, demonstrated the utility of Losartan, an angiotensin receptor blocker (ARB), in slowing aortic growth and preventing dissection, multiple randomized controlled human trials have been unable to consistently reproduce this effect 40, 41, 42. When comparing losartan monotherapy to combination therapy with beta-blocker, two studies have shown slower rates of aneurysm progression in the combination therapy 43, 40. A recent meta-analysis concluded that ARB therapy and in combination with beta-blockade, was capable of slowing the rate of aortic root dilation but did not change the rate of aortic complications and surgery 44. Accordingly, the use of angiotensin receptor blockers (ARBs) may also favorably affect myocardial fibrosis, aortic stiffening, and circulating TGF-beta levels 45. Some experts recommend angiotensin receptor blocker (ARB) as first line therapy, with the addition of beta-blocker as tolerated.
Repair of pectus chest deformity should be postponed until the adolescent growth spurt is completed unless there is cardiopulmonary compromise. Severe scoliosis may require surgical stabilization 46. These operations should be performed at centers with experience managing Marfan patients based on commonly used operative criteria.
Medications
Doctors often prescribe blood pressure lowering drugs to help prevent the aorta from enlarging and to reduce the risk of dissection and rupture. The most commonly used drugs are beta blockers, which cause your heart to beat more slowly and with less force. Beta-blockers are commonly used to treat high blood pressure (hypertension). In this case, beta-blockers help slow down your heart rate and decrease the strength of your heartbeat, which in turn helps to slow down any enlargement of the aorta. However, some people with Marfan syndrome have low blood pressure (hypotension) or have other medical conditions such asthma or depression preventing them from being able to take beta-blockers.
A second class of blood pressure medication called angiotensin receptor blockers (ARBs) is commonly used in the treatment of cardiovascular problems associated with Marfan syndrome 31. This includes medications such as losartan or irbesatan. There is experimental evidence that angiotensin receptor blockers (ARBs) can help by both lowering blood pressure and by blocking TGF-β activity. In animal models of Marfan syndrome, the protective effects of angiotensin receptor blockers (ARBs) was superior to that seen with beta-blockers. In clinical trials, angiotensin receptor blockers (ARBs) have variably been shown to be either better than or as good as beta-blockers in suppressing aneurysm growth, but this may not be true for all patients or in all circumstances. In the largest trial performed to date, young patients receiving beta-blockers at high dosing or angiotensin receptor blockers (ARBs) at standard dosing had a comparable decline in the deviation of the aortic root size from that expected for age and body size (decreasing aortic root Z-score). While both treatments were well tolerated in this study, in general, angiotensin receptor blockers (ARBs) are thought to be better tolerated than beta-blockers. A recent analysis of all of the participants in most of the largest clinical trials for Marfan syndrome (meta-analysis) documented that angiotensin receptor blockers (ARBs) such as losartan or irbesartan can decrease the rate of aortic root growth in individuals with Marfan syndrome. On average, this treatment halved the rate of aortic root growth when compared to individuals who were not receiving medical therapy, with an even greater treatment effect in people that had a documented FBN1 gene variant. It is the stated position of the Marfan Foundation that the choice of treatment should be guided by the particular circumstances. A combination of beta-blocker and angiotensin receptor blocker (ARB) therapy can be considered in circumstances where one or the other type of medication does not achieve an adequate response.
Current American College of Cardiology/American Heart Association guidelines recommend use of either beta blockers or angiotensin receptor blockers (ARBs) or a combination of both at maximally tolerated doses 19. European guidelines recommend use of beta blockers and consider angiotensin receptor blockers (ARBs) as an alternative treatment 20.
Medication Warning
In December 2018, the U.S. Food and Drug Administration (FDA) issued a warning against the use of fluoroquinolone antibiotics (e.g., Avelox, Cipro, Factive, Levaquin, and Ofloxacin) in people with Marfan syndrome and other related genetic aortic conditions 47. The FDA review found that fluoroquinolone antibiotics can increase the occurrence of rare but serious events of ruptures or tears in the main artery of the body, called the aorta. These tears, called aortic dissections, or ruptures of an aortic aneurysm can lead to dangerous bleeding or even death. They can occur with fluoroquinolones for systemic use given by mouth or through an injection. Fluoroquinolones should not be used in patients at increased risk unless there are no other treatment options available. People at increased risk include those with a history of blockages or aneurysms (abnormal bulges) of the aorta or other blood vessels, high blood pressure, certain genetic disorders that involve blood vessel changes, and the elderly 47.
Skeletal problems
Skeletal problems that develop as a result of Marfan syndrome can sometimes cause significant pain and discomfort.
They may also affect your appearance, which some people find affects their confidence and self-esteem.
There are a number of ways skeletal symptoms can be treated. Some of these are outlined below.
Loose, painful joints
Seventy per cent of people with Marfan syndrome have pain in and around their joints. Good posture, exercises, and the use of joint supports, as well as pain relief, such as paracetamol and non-steroidal anti-inflammatory drugs (NSAIDs), can prove helpful.
Scoliosis
Treatment for curvature of the spine (scoliosis) will depend on how severely your spine is curved. If your spine is mildly curved, your treatment team will closely monitor it to see whether it gets worse.
In some cases, particularly in children who are still growing, a back brace may be recommended. The brace won’t cure scoliosis, but it may stop it getting worse.
A back brace usually needs to be worn for 23 hours a day, and is only removed for baths, showers, swimming, and contact sports. Some children find wearing a back brace difficult at first, but it needs to be worn for the correct amount of time to be effective.
Surgery will usually be needed to straighten your spine if it curves by 40 degrees or more. Straightening the spine will help alleviate problems such as restricted breathing and back pain.
A number of different types of surgery can be used to treat scoliosis. The type recommended will depend on your age and individual circumstances.
In young children – generally those under the age of 10 – growing rods are inserted, which allow for continued growth while partially correcting the curvature of the spine.
In teenagers and young adults, an operation called spinal fusion may be carried out. This is where the spine is straightened using metal rods that are attached with screws, hooks, and wires. Bone grafts are used to fuse the spine in place.
Surgery for adults with scoliosis is usually only recommended if the spinal curvature is severe, getting significantly worse, or the nerves in the spine are being compressed.
The two main types of surgery used are decompression surgery, where the disc or bone pressing on a nerve is removed, and spinal fusion surgery.
These are major operations that can take a year or more to fully recover from. They also carry a risk of potentially serious complications, such as infection, blood clots, and, in rare cases, nerve damage.
Convex and concave chest
Marfan syndrome can sometimes affect the natural position of the chest. Your chest is concave if it caves inwards, and convex if it protrudes outwards.
In rare cases, a person’s chest can be severely concave and press against their lungs, affecting breathing. Surgery will usually be required to help ease the pressure on the lungs.
Surgery for a concave chest involves raising the breastbone (sternum) and ribs, and fixing them in place with a metal bar. Once the breastbone and ribs are fixed in position, the bar will be removed.
A convex chest shouldn’t cause any health problems and won’t usually require treatment. However, some people with a convex chest choose to have treatment for cosmetic reasons.
Physiotherapy
Physiotherapy uses physical methods such as exercise, massage, and manipulation to promote healing and wellbeing. It can help improve your range of movement and strengthen muscle support.
If skeletal problems are making it difficult for you to get around, physiotherapy may help make moving easier and more comfortable.
Heart problems
Marfan syndrome can cause serious heart problems, which can be fatal. It’s therefore important that your heart is treated as a priority.
You’ll need to have regular check-ups with a cardiologist, who will be able to monitor your heart. This may mean having a yearly echocardiogram, where an ultrasound scan produces an image of your heart.
An echocardiogram can identify the structure, thickness, and movement of the aorta and each heart valve, enabling any potential heart-related complications to be detected and treated as soon as possible.
The 2010 American College of Cardiology, American Heart Association and American Association for Thoracic Surgery (ACC/AHA/AATS) thoracic aorta guidelines recommended that echocardiography is performed at initial diagnosis and 6 months to assess the aortic root and ascending aorta in patients with Marfan syndrome 48. A 6-month echocardiogram is performed to confirm the stability of the aortic dimension. Nomograms and Z-scores are used to identify aortic dilatation because the normal range for aortic diameter varies with body size and age. Undiagnosed and untreated Marfan syndrome is often associated with aortic dissection that begins above the coronary ostia and can extend the entire length of the aorta, known as a type 1 dissection in the DeBakey classification. Based on a review from the International Registry of Aortic Dissection (IRAD) 49, Marfan syndrome causes an aortic dissection in 50% of those under age 40, compared to only 2% of older patients with aortic dissection and no patients over age 70 50.
Aortic dissection and rupture are preventable in patients with Marfan syndrome by replacement of the ascending aorta. Preventive (prophylactic) surgery is recommended when the ascending aorta’s diameter at the aortic sinuses’ level reaches 5.0 cm 2. Patients with an aortic diameter of less than 2.75 cm/m² are considered to be at low risk of aortic dissection, those with 2.75 to 4.24 cm/m2 are at moderate risk, and those with greater than 4.25 cm/m² are at high risk 2. A family history of aortic dissection, increased rate of aortic dilation (greater than 2 mm per year), severe aortic valve regurgitation with left ventricular dilation, and relative feasibility of aortic valve-sparing surgery are also indicators for early surgical intervention 51.
Mitral valve prolapse (MVP) is identified in 40% to 54% of patients with Marfan syndrome 52, 53. Since it is nonspecific, only 1 point in the systemic score is assigned for mitral valve prolapse 54. The frequency of mitral valve prolapse in Marfan syndrome increases with age and is more prevalent in women. Tricuspid valve prolapse may also occur. Patients with Marfan syndrome and mitral valve prolapse have mild regurgitation 52. In these patients, the worsening of mild regurgitation is due to spontaneous rupture of the chordae tendineae or infective endocarditis. Heart failure caused by mitral valve prolapse and mild regurgitation represents a major source of morbidity and mortality in young children with rapidly progressive Marfan syndrome. Due to disrupted granular fibers from the proximal aorta to the base of the innominate artery, care should be taken without cross-clamp placement for patients with Marfan syndrome with mitral valve prolapse 55. The presence of aortic valve regurgitation should be taken into account when performing cardioplegia to ensure adequate myocardial protection. Freedom from moderate or greater mitral regurgitation at 5, 10, and 20 years is about 95%, 89%, and 69%, respectively 56. Patients with Marfan syndrome may have cardiomyopathy (a disease of the heart muscle that makes it harder for the heart to pump blood to the rest of the body) with biventricular enlargement and mild systolic dysfunction 57. Patients may exhibit proximal ascending aortic dilation, proximal main pulmonary artery dilation, thickening and prolapse of the atrioventricular valves, and mitral annular calcification.
Some of the possible treatment options for the heart are described below.
Surgery
If your cardiologist feels it’s necessary, you may need to have heart surgery to reduce your risk of developing life-threatening complications.
The most common type of heart surgery carried out on people with Marfan syndrome is an operation to replace a section of an enlarged aorta. This operation must be carried out before the aorta becomes too big.
You’ll have an echocardiogram every year to monitor the diameter of the aorta. Surgery will be considered when it measures between 4.5cm and 4.8cm (about 1.8-1.9 inches).
If your aorta is severely enlarged, the risk of it tearing or splitting (rupturing) during the operation will be too high for the benefits to outweigh the risks. Emergency surgery will be needed if your aorta ruptures or tears.
Eye problems
If you’ve been diagnosed with Marfan syndrome, you may be referred to an ophthalmologist, who will assess your eyes and vision.
You may also need to have an annual check-up to help identify any new developments. Eye problems associated with Marfan syndrome are potentially serious and may lead to a permanent loss of vision. The vision problems associated with a dislocated lens in your eye often can be corrected with glasses or contact lenses.
Some of the treatment options for eye problems are outlined below.
Cataracts
If you develop cataracts as a result of Marfan syndrome, you may need surgery to replace the clouded lens with an artificial one.
Cataract surgery is usually performed as keyhole surgery, through a very small cut, under local anesthetic.
Glaucoma
People with Marfan syndrome have a higher risk of developing glaucoma, a condition caused by increased pressure in the eyeball.
Once glaucoma has caused vision loss, it can’t be cured. Your eyes will therefore be carefully monitored to detect any signs of the condition.
Although glaucoma can’t be cured, it’s usually possible to prevent it getting worse. Treatment options include eye drops, laser treatment, or surgery.
Glasses and contact lenses
If you’re short-sighted, your vision can usually be corrected using glasses or contact lenses.
If the transparent structure at the front of your eye (the lens) is dislocated, specially designed glasses or contact lenses can sometimes be used to bend (refract) light around the dislocated lens.
In rare cases where a person’s vision is significantly affected, the lens may need to be replaced with an artificial one.
Surgical and other procedures
Depending upon your signs and symptoms, procedures might include:
- Aortic repair. If your aorta’s diameter enlarges quickly or reaches about 2 inches (5 centimeters), your doctor may recommend an operation to replace a portion of your aorta with a tube made of synthetic material. This can help prevent a life-threatening rupture. Your aortic valve may need to be replaced as well.
- Scoliosis treatment. For some children and adolescents, doctors recommend a custom-made back brace, which is worn nearly continuously until growth is complete. If the curve in your child’s spine is too great, your doctor may suggest surgery to straighten the spine.
- Breastbone corrections. Surgical options are available to correct the appearance of a sunken or protruding breastbone. Because these operations are often considered to be for cosmetic purposes, your insurance might not cover the costs.
- Eye surgeries. If parts of your retina have torn or come loose from the back of your eye, surgical repair is usually successful. If you have cataracts, your clouded lens can be replaced with an artificial lens.
Marfan syndrome in Pregnancy Treatment
Pregnancy with Marfan syndrome needs to be managed by a high-risk obstetrician, preferably at a medical center with expertise in aortic surgery and management of Marfan syndrome patients.
Pregnancy is recognized as a risk factor for aortic dissection in women in the general population and in women with Marfan syndrome 20, 48, 58. The risk for aortic dissection is reported both during pregnancy, primarily during the third trimester, and up to several months postpartum 59, 60, 58. However, pregnancy-associated dissections in the general population are rare 61, 62. In women with Marfan syndrome, the risk of dissection is associated with an increased aortic root dilatation rate in pregnancy 63. In the majority of reported aortic dissections, the diagnosis of Marfan syndrome had not been made prior to the aortic dissection 58. Therefore, a missed diagnosis of Marfan syndrome is an important risk factor for pregnancy-associated aortic dissections and underscores the crucial role for diagnosis and genetic testing of family members 59.
Importantly, women with Marfan syndrome should have genetic counselling and a cardiovascular assessment prior to pregnancy. Aortic diameter remains a risk factor for dissection, and current recommendations for the care of women with Marfan syndrome who are considering conceiving are based on aortic root diameter. There are important considerations regarding prophylactic surgery and the risk for Stanford Type B dissection (an aortic dissection that starts in the descending aorta, beyond the origin of the left subclavian artery, and does not involve the ascending aorta). Stanford Type B dissection is classified as “complicated” if there are signs like rupture, malperfusion, or severe pain, which usually require urgent treatment with endovascular repair or open surgery 64. Otherwise, Type B Aortic Dissection is treated with medication and careful blood pressure control, with a follow-up plan for potential future interventions 64. Type B Aortic Dissections associated with pregnancy may occur without substantial aortic dilation 58, 60.
Stanford Type A aortic dissection is a serious medical condition requiring immediate surgical intervention. It involves a tear in the inner lining of the aorta, specifically the ascending aorta, which can extend to the aortic arch and beyond. This type of dissection is a surgical emergency due to the high risk of complications like cardiac tamponade, aortic rupture, or stroke. The risk of pregnancy-associated type A dissection is considered low when maximal diameter of the aortic root is < 4.0 cm, provided beta-adrenergic receptor blockers are used throughout the pregnancy and the postpartum period 6. When the aortic root diameter is > 4.5 cm, prophylactic valve-sparing aortic root replacement surgery should be considered before the pregnancy 65. When the diameter is between 4.0 and 4.5 cm, the decision is made on a case-by-case basis. Importantly, aortic root replacement does not completely eliminate the risk of aortic dissections during preganacy 60, 66.
Vaginal delivery is possible when aortic root diameter is < 4.0 cm, and, in addition to the usual indications, C-section should be mainly considered , when the aortic diameter is > 4.5 cm. In some cases, elective delivery before full term is recommended to lessen hemodynamic stresses, and an expedited second stage of labor with the use of regional anesthesia should be considered to prevent blood pressure spikes 65. Lastly, although beta-adrenergic receptor blockers are compatible with pregnancy but angiotensin-receptor blockers (ARBs) should not be used during pregnancy because of potential harm to the developing fetus 67. The negative effects of Warfarin on fetal development, and the risk for dissection associated with lactation following delivery are also key concerns.
Coping and support
Living with a genetic disorder can be extremely difficult for both adults and children. Adults who receive a diagnosis later in life may wonder how the disease will affect their careers, their relationships and their sense of themselves. And they may worry about passing the defective gene to their children.
But Marfan syndrome can be even harder on young people, especially because the often-inherent self-consciousness of childhood and adolescence may be exacerbated by the disease’s effect on appearance, academic performance and motor skills.
Psychological support
Being diagnosed with Marfan syndrome can sometimes be difficult to deal with emotionally. If your child has been diagnosed with the syndrome, you may be worried or upset about how it will affect them.
Speak to your doctor if you or your child are finding the diagnosis difficult to cope with. They may be able to put you in touch with a support group through the Marfan Foundation (http://www.marfan.org/), or refer you to a counseling service.
Young people with Marfan syndrome may develop low self-esteem because of their physical appearance. As the symptoms tend to be most apparent during the teenage years, a young person may find them difficult to deal with. Speak to your doctor if you’re concerned.
Working together, parents, teachers and medical professionals can provide children with both emotional support and practical solutions for some of the more distressing aspects of the disease. For example, children with Marfan syndrome may struggle in school because of eye problems that can be corrected with glasses or contact lenses.
For most young people, cosmetic concerns are at least as important as academic ones. Parents can help by anticipating these concerns and offering solutions:
- Contact lenses instead of glasses
- A brace for scoliosis
- Dental work for crowded teeth
- Clothes that flatter a tall, thin frame
In the long run, accurate information about the disease, good medical care and strong social support can help both children and adults cope with Marfan syndrome.
Lifestyle
It’s not usually necessary to make significant lifestyle changes if you have Marfan syndrome. However, a young person’s career choice may be restricted.
Keeping fit through regular moderate exercise and eating a healthy, balanced diet will help improve your overall health.
Sport
If you have Marfan syndrome, you may be advised to avoid certain sports. For example, some people may not be able to participate in contact sports like rugby.
Other activities that may need to be avoided include:
- long-distance running
- heavy weightlifting
- gymnastics
- climbing
These types of sporting activities can place a strain on your heart. They raise your blood pressure and heart rate, which may increase the risk of an aortic tear.
These activities also place a strain on your joints. As people with Marfan syndrome often have weak joints, their risk of sustaining a joint injury during these activities may be increased.
Your cardiologist will be able to give you more advice about which sports and physical activities are suitable for you.
Marfan syndrome life expectancy
The prognosis (outlook) of Marfan syndrome largely depends on the severity of the complications previously mentioned and the degree of progressive aortic dilation which can lead to death at a young age. However, the life expectancy for patients with Marfan syndrome has improved over time, presumably due to improved detection and intervention, including surgical procedures and the use of beta-blockers. The lifespan of untreated patients with the classic Marfan syndrome was approximately 32 years in 1972 but the average lifespan is now approximately 72 years 68, 69. Marfan syndrome life expectancy is now almost similar to persons without Marfan syndrome. Life expectancy is significantly lower in men than in women. A Norwegian study by Vanem et al 70 found that out of 84 adults with Marfan syndrome first investigated in 2003-2004, 16 (19.0%) were deceased by 2014-2015 follow-up, including 11 (68.8%) who had died of cardiovascular causes. Standardized mortality ratios were 8.20 for men and 3.85 for women. However, cardiovascular impairment is still the most common cause of mortality, mainly because of sudden death in an undiagnosed patient and in a newly diagnosed patient whose disease process has worsened beyond the scope of medical or surgical cure. A study by Reis et al 71 of patients with Marfan syndrome found that those who had an aortic root diameter of 45 mm or more at the time of diagnosis had a significantly worse survival rate over the study’s mean 12.4-year follow-up period.
Cardiovascular disease (aortic dilatation and dissection) is the major cause of sickness and death in Marfan syndrome 72. Without proper medical management, Marfan syndrome can be lethal in young adulthood, with death occurring at an average age of 30 to 40 years 73. With appropriate medical and surgical care, however, patients with Marfan syndrome can expect a lifespan approaching normal 15.
Infant morbidity, as related to cardiovascular disease, is due to progression of mitral valve prolapse to mitral regurgitation and often occurs in conjunction with tricuspid prolapse and regurgitation 72. Progression to congestive heart failure is a leading cause of cardiovascular morbidity and mortality, as well as the leading indicator for cardiovascular surgery 72.
Death later in life is usually due to chronic aortic regurgitation and ascending aortic dissection. Aortic root dissection is uncommon in childhood and adolescence 72.
- Dietz H. FBN1-Related Marfan Syndrome. 2001 Apr 18 [Updated 2022 Feb 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1335[↩][↩][↩]
- Salik I, Rawla P. Marfan Syndrome. [Updated 2023 Jan 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537339[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Marfan syndrome. https://medlineplus.gov/genetics/condition/marfan-syndrome[↩][↩][↩][↩][↩][↩]
- Marfan Syndrome. https://rarediseases.org/rare-diseases/marfan-syndrome[↩]
- Marfan Syndrome. https://marfan.org/conditions/marfan-syndrome[↩]
- Milewicz DM, Braverman AC, De Backer J, Morris SA, Boileau C, Maumenee IH, Jondeau G, Evangelista A, Pyeritz RE. Marfan syndrome. Nat Rev Dis Primers. 2021 Sep 2;7(1):64. doi: 10.1038/s41572-021-00298-7. Erratum in: Nat Rev Dis Primers. 2022 Jan 17;8(1):3. doi: 10.1038/s41572-022-00338-w[↩][↩]
- Muiño-Mosquera L, Cervi E, De Groote K, Dewals W, Fejzic Z, Kazamia K, Mathur S, Milleron O, Mir TS, Nielsen DG, Odermarsky M, Sabate-Rotes A, van der Hulst A, Valenzuela I, Jondeau G. Management of aortic disease in children with FBN1-related Marfan syndrome. Eur Heart J. 2024 Oct 14;45(39):4156-4169. doi: 10.1093/eurheartj/ehae526[↩]
- Edouard T, Picot MC, Bajanca F, Huguet H, Guitarte A, Langeois M, Chesneau B, Van Kien PK, Garrigue E, Dulac Y, Amedro P. Health-related quality of life in children and adolescents with Marfan syndrome or related disorders: a controlled cross-sectional study. Orphanet J Rare Dis. 2024 Apr 30;19(1):180. doi: 10.1186/s13023-024-03191-0[↩]
- Taniguchi Y, Takeda N, Inuzuka R, Matsubayashi Y, Kato S, Doi T, Yagi H, Yamauchi H, Ando M, Oshima Y, Tanaka S. Impact of pathogenic FBN1 variant types on the development of severe scoliosis in patients with Marfan syndrome. J Med Genet. 2023 Jan;60(1):74-80. doi: 10.1136/jmedgenet-2021-108186[↩]
- Laganà G, Venza N, Malara A, Liguori C, Cozza P, Pisano C. Obstructive Sleep Apnea, Palatal Morphology, and Aortic Dilatation in Marfan Syndrome Growing Subjects: A Retrospective Study. Int J Environ Res Public Health. 2021 Mar 16;18(6):3045. doi: 10.3390/ijerph18063045[↩]
- Trawicka A, Lewandowska-Walter A, Majkowicz M, Sabiniewicz R, Woźniak-Mielczarek L. Health-Related Quality of Life of Patients with Marfan Syndrome-Polish Study. Int J Environ Res Public Health. 2022 Jun 2;19(11):6827. doi: 10.3390/ijerph19116827[↩]
- Pollock L, Ridout A, Teh J, Nnadi C, Stavroulias D, Pitcher A, Blair E, Wordsworth P, Vincent TL. The Musculoskeletal Manifestations of Marfan Syndrome: Diagnosis, Impact, and Management. Curr Rheumatol Rep. 2021 Nov 26;23(11):81. doi: 10.1007/s11926-021-01045-3[↩]
- Du Q, Zhang D, Zhuang Y, Xia Q, Wen T, Jia H. The Molecular Genetics of Marfan Syndrome. Int J Med Sci. 2021 May 27;18(13):2752-2766. doi: 10.7150/ijms.60685[↩]
- Baban A, Parlapiano G, Cicenia M, Armando M, Franceschini A, Pacifico C, Panfili A, Zinzanella G, Romanzo A, Fusco A, Caiazza M, Perri G, Galletti L, Digilio MC, Buonuomo PS, Bartuli A, Novelli A, Raponi M, Limongelli G. Unique Features of Cardiovascular Involvement and Progression in Children with Marfan Syndrome Justify Dedicated Multidisciplinary Care. J Cardiovasc Dev Dis. 2024 Apr 3;11(4):114. doi: 10.3390/jcdd11040114[↩]
- Zeigler SM, Sloan B, Jones JA. Pathophysiology and Pathogenesis of Marfan Syndrome. Adv Exp Med Biol. 2021;1348:185-206. doi: 10.1007/978-3-030-80614-9_8[↩][↩]
- Andersen NH, Hauge EM, Baad-Hansen T, Groth KA, Berglund A, Gravholt CH, Stochholm K. Musculoskeletal diseases in Marfan syndrome: a nationwide registry study. Orphanet J Rare Dis. 2022 Mar 5;17(1):118. doi: 10.1186/s13023-022-02272-2[↩]
- Zodanu GKE, Hwang JH, Mehta Z, Sisniega C, Barsegian A, Kang X, Biniwale R, Si MS, Satou GM, Halnon N, Ucla Congenital Heart Defect BioCore Faculty, Grody WW, Van Arsdell GS, Nelson SF, Touma M. High-Throughput Genomics Identify Novel FBN1/2 Variants in Severe Neonatal Marfan Syndrome and Congenital Heart Defects. Int J Mol Sci. 2024 May 17;25(10):5469. doi: 10.3390/ijms25105469[↩]
- von Kodolitsch Y, Robinson PN. Marfan syndrome: an update of genetics, medical and surgical management. Heart. 2007 Jun;93(6):755-60. doi: 10.1136/hrt.2006.098798[↩]
- Isselbacher EM, Preventza O, Hamilton Black J, Augoustides JG, Beck AW, Bolen MA et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease: a report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. Circulation 2022;146:e334–482. 10.1161/CIR.0000000000001106[↩][↩]
- Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, Evangelista A, Falk V, Frank H, Gaemperli O, Grabenwöger M, Haverich A, Iung B, Manolis AJ, Meijboom F, Nienaber CA, Roffi M, Rousseau H, Sechtem U, Sirnes PA, Allmen RS, Vrints CJ; ESC Committee for Practice Guidelines. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J. 2014 Nov 1;35(41):2873-926. doi: 10.1093/eurheartj/ehu281. Epub 2014 Aug 29. Erratum in: Eur Heart J. 2015 Nov 1;36(41):2779. doi: 10.1093/eurheartj/ehv178[↩][↩][↩]
- Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005 Nov 15;139C(1):4-9. doi: 10.1002/ajmg.c.30068[↩]
- Hilhorst-Hofstee Y, Rijlaarsdam ME, Scholte AJ, Swart-van den Berg M, Versteegh MI, van der Schoot-van Velzen I, Schäbitz HJ, Bijlsma EK, Baars MJ, Kerstjens-Frederikse WS, Giltay JC, Hamel BC, Breuning MH, Pals G. The clinical spectrum of missense mutations of the first aspartic acid of cbEGF-like domains in fibrillin-1 including a recessive family. Hum Mutat. 2010 Dec;31(12):E1915-27. doi: 10.1002/humu.21372[↩][↩][↩][↩]
- Sakai LY, Keene DR, Glanville RW, Bächinger HP. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J Biol Chem. 1991 Aug 5;266(22):14763-70. https://www.jbc.org/article/S0021-9258(18)98752-1/pdf[↩][↩]
- Dietz HC, Ramirez F, Sakai LY. Marfan’s syndrome and other microfibrillar diseases. Adv Hum Genet. 1994;22:153-86. doi: 10.1007/978-1-4757-9062-7_4[↩][↩]
- FBN1 gene. https://medlineplus.gov/genetics/gene/fbn1[↩][↩]
- Reinhardt DP, Keene DR, Corson GM, Pöschl E, Bächinger HP, Gambee JE, Sakai LY. Fibrillin-1: organization in microfibrils and structural properties. J Mol Biol. 1996 Apr 26;258(1):104-16. doi: 10.1006/jmbi.1996.0237[↩][↩]
- Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol. 1995 May;129(4):1165-76. https://pmc.ncbi.nlm.nih.gov/articles/instance/2120487/pdf/jc12941165.pdf[↩][↩]
- Ammash NM, Sundt TM, Connolly HM. Marfan syndrome-diagnosis and management. Curr Probl Cardiol. 2008 Jan;33(1):7-39. doi: 10.1016/j.cpcardiol.2007.10.001[↩]
- LeMaire SA, Russell L. Epidemiology of thoracic aortic dissection. Nat Rev Cardiol. 2011 Feb;8(2):103-13. doi: 10.1038/nrcardio.2010.187[↩]
- Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006 Apr 7;312(5770):117-21. doi: 10.1126/science.1124287[↩]
- Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med. 2008 Jun 26;358(26):2787-95. doi: 10.1056/NEJMoa0706585[↩][↩][↩]
- Chiu HH, Wu MH, Chen HC, Kao FY, Huang SK. Epidemiological profile of Marfan syndrome in a general population: a national database study. Mayo Clin Proc. 2014 Jan;89(1):34-42. doi: 10.1016/j.mayocp.2013.08.022[↩]
- Peng Q, Deng Y, Yang Y, Liu H. A novel fibrillin-1 gene missense mutation associated with neonatal Marfan syndrome: a case report and review of the mutation spectrum. BMC Pediatr. 2016 Apr 30;16:60. doi: 10.1186/s12887-016-0598-6[↩]
- De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996 Apr 24;62(4):417-26. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R[↩]
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. https://jmg.bmj.com/content/47/7/476.long[↩][↩][↩][↩][↩][↩]
- Stuart AG, Williams A. Marfan’s syndrome and the heart. Arch Dis Child. 2007 Apr;92(4):351-6. doi: 10.1136/adc.2006.097469[↩]
- Lindsay ME. Medical management of aortic disease in children with Marfan syndrome. Curr Opin Pediatr. 2018 Oct;30(5):639-644. doi: 10.1097/MOP.0000000000000671[↩][↩][↩]
- Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005 Dec 3;366(9501):1965-76. doi: 10.1016/S0140-6736(05)67789-6[↩]
- Salim MA, Alpert BS, Ward JC, Pyeritz RE. Effect of beta-adrenergic blockade on aortic root rate of dilation in the Marfan syndrome. Am J Cardiol. 1994 Sep 15;74(6):629-33. doi: 10.1016/0002-9149(94)90762-5[↩]
- Forteza A, Evangelista A, Sánchez V, Teixidó-Turà G, Sanz P, Gutiérrez L, Gracia T, Centeno J, Rodríguez-Palomares J, Rufilanchas JJ, Cortina J, Ferreira-González I, García-Dorado D. Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur Heart J. 2016 Mar 21;37(12):978-85. doi: 10.1093/eurheartj/ehv575[↩][↩]
- Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L; Pediatric Heart Network Investigators. Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014 Nov 27;371(22):2061-71. doi: 10.1056/NEJMoa1404731[↩]
- Milleron O, Arnoult F, Ropers J, Aegerter P, Detaint D, Delorme G, Attias D, Tubach F, Dupuis-Girod S, Plauchu H, Barthelet M, Sassolas F, Pangaud N, Naudion S, Thomas-Chabaneix J, Dulac Y, Edouard T, Wolf JE, Faivre L, Odent S, Basquin A, Habib G, Collignon P, Boileau C, Jondeau G. Marfan Sartan: a randomized, double-blind, placebo-controlled trial. Eur Heart J. 2015 Aug 21;36(32):2160-6. doi: 10.1093/eurheartj/ehv151[↩]
- Mullen M, Jin XY, Child A, Stuart AG, Dodd M, Aragon-Martin JA, Gaze D, Kiotsekoglou A, Yuan L, Hu J, Foley C, Van Dyck L, Knight R, Clayton T, Swan L, Thomson JDR, Erdem G, Crossman D, Flather M; AIMS Investigators. Irbesartan in Marfan syndrome (AIMS): a double-blind, placebo-controlled randomised trial. Lancet. 2019 Dec 21;394(10216):2263-2270. doi: 10.1016/S0140-6736(19)32518-8[↩]
- Al-Abcha A, Saleh Y, Mujer M, Boumegouas M, Herzallah K, Charles L, Elkhatib L, Abdelkarim O, Kehdi M, Abela GS. Meta-analysis Examining the Usefulness of Angiotensin Receptor blockers for the Prevention of Aortic Root Dilation in Patients With the Marfan Syndrome. Am J Cardiol. 2020 Aug 1;128:101-106. doi: 10.1016/j.amjcard.2020.04.034[↩]
- Karur GR, Pagano JJ, Bradley T, Lam CZ, Seed M, Yoo SJ, Grosse-Wortmann L. Diffuse Myocardial Fibrosis in Children and Adolescents With Marfan Syndrome and Loeys-Dietz Syndrome. J Am Coll Cardiol. 2018 Oct 30;72(18):2279-2281. doi: 10.1016/j.jacc.2018.07.095[↩]
- Jones KB, Erkula G, Sponseller PD, Dormans JP. Spine deformity correction in Marfan syndrome. Spine (Phila Pa 1976). 2002 Sep 15;27(18):2003-12. doi: 10.1097/00007632-200209150-00008[↩]
- FDA warns about increased risk of ruptures or tears in the aorta blood vessel with fluoroquinolone antibiotics in certain patients. https://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-increased-risk-ruptures-or-tears-aorta-blood-vessel-fluoroquinolone-antibiotics[↩][↩]
- Hiratzka LF, Bakris GL, Beckman JA, et al. American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American College of Radiology; American Stroke Association; Society of Cardiovascular Anesthesiologists; Society for Cardiovascular Angiography and Interventions; Society of Interventional Radiology; Society of Thoracic Surgeons; Society for Vascular Medicine. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010 Apr 6;121(13):e266-369. doi: 10.1161/CIR.0b013e3181d4739e. Epub 2010 Mar 16. Erratum in: Circulation. 2010 Jul 27;122(4):e410.[↩][↩]
- Januzzi JL, Isselbacher EM, Fattori R, Cooper JV, Smith DE, Fang J, Eagle KA, Mehta RH, Nienaber CA, Pape LA; International Registry of Aortic Dissection (IRAD). Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004 Feb 18;43(4):665-9. doi: 10.1016/j.jacc.2003.08.054[↩]
- Mehta RH, O’Gara PT, Bossone E, Nienaber CA, Myrmel T, Cooper JV, Smith DE, Armstrong WF, Isselbacher EM, Pape LA, Eagle KA, Gilon D; International Registry of Acute Aortic Dissection (IRAD) Investigators. Acute type A aortic dissection in the elderly: clinical characteristics, management, and outcomes in the current era. J Am Coll Cardiol. 2002 Aug 21;40(4):685-92. doi: 10.1016/s0735-1097(02)02005-3[↩]
- Svensson LG, Adams DH, Bonow RO, et al. Aortic valve and ascending aorta guidelines for management and quality measures: executive summary. Ann Thorac Surg. 2013 Apr;95(4):1491-505. doi: 10.1016/j.athoracsur.2012.12.027[↩]
- Rybczynski M, Mir TS, Sheikhzadeh S, Bernhardt AM, Schad C, Treede H, Veldhoen S, Groene EF, Kühne K, Koschyk D, Robinson PN, Berger J, Reichenspurner H, Meinertz T, von Kodolitsch Y. Frequency and age-related course of mitral valve dysfunction in the Marfan syndrome. Am J Cardiol. 2010 Oct 1;106(7):1048-53. doi: 10.1016/j.amjcard.2010.05.038[↩][↩]
- Faivre L, Collod-Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007 Sep;81(3):454-66. doi: 10.1086/520125[↩]
- Roman MJ, Devereux RB, Kramer-Fox R, Spitzer MC. Comparison of cardiovascular and skeletal features of primary mitral valve prolapse and Marfan syndrome. Am J Cardiol. 1989 Feb 1;63(5):317-21. doi: 10.1016/0002-9149(89)90338-x[↩]
- Moon J, Shin MS, Lee HJ, Chung WJ, Park CH, Park KY. Newly developed aortic dissection after aorta cannulation during mitral valve surgery in a patient with marfan syndrome. Korean Circ J. 2012 Jun;42(6):437-40. doi: 10.4070/kcj.2012.42.6.437[↩]
- David TE, Armstrong S, McCrindle BW, Manlhiot C. Late outcomes of mitral valve repair for mitral regurgitation due to degenerative disease. Circulation. 2013 Apr 9;127(14):1485-92. doi: 10.1161/CIRCULATIONAHA.112.000699[↩]
- Alpendurada F, Wong J, Kiotsekoglou A, Banya W, Child A, Prasad SK, Pennell DJ, Mohiaddin RH. Evidence for Marfan cardiomyopathy. Eur J Heart Fail. 2010 Oct;12(10):1085-91. doi: 10.1093/eurjhf/hfq127[↩]
- Braverman AC, Mittauer E, Harris KM, Evangelista A, Pyeritz RE, Brinster D, Conklin L, Suzuki T, Fanola C, Ouzounian M, Chen E, Myrmel T, Bekeredjian R, Hutchison S, Coselli J, Gilon D, O’Gara P, Davis M, Isselbacher E, Eagle K. Clinical Features and Outcomes of Pregnancy-Related Acute Aortic Dissection. JAMA Cardiol. 2021 Jan 1;6(1):58-66. doi: 10.1001/jamacardio.2020.4876[↩][↩][↩][↩]
- Roman MJ, Pugh NL, Hendershot TP, Devereux RB, et al. Aortic Complications Associated With Pregnancy in Marfan Syndrome: The NHLBI National Registry of Genetically Triggered Thoracic Aortic Aneurysms and Cardiovascular Conditions (GenTAC). J Am Heart Assoc. 2016 Aug 11;5(8):e004052. doi: 10.1161/JAHA.116.004052[↩][↩]
- Cauldwell M, Steer PJ, Curtis SL, Mohan A, Dockree S, Mackillop L, Parry HM, Oliver J, Sterrenberg M, Wallace S, Malin G, Partridge G, Freeman LJ, Bolger AP, Siddiqui F, Wilson D, Simpson M, Walker N, Hodson K, Thomas K, Bredaki F, Mercaldi R, Walker F, Johnson MR. Maternal and fetal outcomes in pregnancies complicated by Marfan syndrome. Heart. 2019 Nov;105(22):1725-1731. doi: 10.1136/heartjnl-2019-314817[↩][↩][↩]
- Banerjee A, Begaj I, Thorne S. Aortic dissection in pregnancy in England: an incidence study using linked national databases. BMJ Open. 2015 Aug 20;5(8):e008318. doi: 10.1136/bmjopen-2015-008318[↩]
- Meijboom LJ, Vos FE, Timmermans J, Boers GH, Zwinderman AH, Mulder BJ. Pregnancy and aortic root growth in the Marfan syndrome: a prospective study. Eur Heart J. 2005 May;26(9):914-20. doi: 10.1093/eurheartj/ehi103[↩]
- Pacini L, Digne F, Boumendil A, Muti C, Detaint D, Boileau C, Jondeau G. Maternal complication of pregnancy in Marfan syndrome. Int J Cardiol. 2009 Aug 14;136(2):156-61. doi: 10.1016/j.ijcard.2008.04.035[↩]
- Thakkar D, Dake MD. Management of Type B Aortic Dissections: Treatment of Acute Dissections and Acute Complications from Chronic Dissections. Tech Vasc Interv Radiol. 2018 Sep;21(3):124-130. doi: 10.1053/j.tvir.2018.06.001[↩][↩]
- Regitz-Zagrosek V, Roos-Hesselink JW, Bauersachs J, Blomström-Lundqvist C, Cífková R, De Bonis M, Iung B, Johnson MR, Kintscher U, Kranke P, Lang IM, Morais J, Pieper PG, Presbitero P, Price S, Rosano GMC, Seeland U, Simoncini T, Swan L, Warnes CA; ESC Scientific Document Group. 2018 ESC Guidelines for the management of cardiovascular diseases during pregnancy. Eur Heart J. 2018 Sep 7;39(34):3165-3241. doi: 10.1093/eurheartj/ehy340[↩][↩]
- Williams D et al. Pregnancy after Aortic Root Replacement in Marfan’s Syndrome: A Case Series and Review of the Literature. AJP Rep 8, e234–e240, doi: 10.1055/s-0038-1675347[↩]
- Bullo M, Tschumi S, Bucher BS, Bianchetti MG & Simonetti GD Pregnancy outcome following exposure to angiotensin-converting enzyme inhibitors or angiotensin receptor antagonists: a systematic review. Hypertension 60, 444–450, doi: 10.1161/HYPERTENSIONAHA.112.196352[↩]
- Finkbohner R, Johnston D, Crawford ES, Coselli J, Milewicz DM. Marfan syndrome. Long-term survival and complications after aortic aneurysm repair. Circulation. 1995 Feb 1;91(3):728-33. doi: 10.1161/01.cir.91.3.728[↩]
- Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, Boxer M, Devereux RB, Tsipouras P. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995 Jan 15;75(2):157-60. doi: 10.1016/s0002-9149(00)80066-1[↩]
- Vanem TT, Geiran OR, Krohg-Sørensen K, Røe C, Paus B, Rand-Hendriksen S. Survival, causes of death, and cardiovascular events in patients with Marfan syndrome. Mol Genet Genomic Med. 2018 Nov;6(6):1114-1123. doi: 10.1002/mgg3.489[↩]
- Reis JF, Mano TB, Rito T, Branco LM, Fragata J, Martins JD, Ferreira RC, Sousa L. Long term follow-up of Marfan Syndrome – experience of an adult congenital heart disease centre. Am J Cardiovasc Dis. 2022 Apr 15;12(2):92-101. https://pmc.ncbi.nlm.nih.gov/articles/PMC9123413[↩]
- Genetics of Marfan Syndrome. https://emedicine.medscape.com/article/946315-overview#showall[↩][↩][↩][↩]
- Goldfinger JZ, Preiss LR, Devereux RB, Roman MJ, Hendershot TP, Kroner BL, Eagle KA; GenTAC Registry Consortium. Marfan Syndrome and Quality of Life in the GenTAC Registry. J Am Coll Cardiol. 2017 Jun 13;69(23):2821-2830. doi: 10.1016/j.jacc.2017.04.026[↩]





{kind=link}