Contents
- Restrictive cardiomyopathy
- Restrictive cardiomyopathy causes
- Restrictive cardiomyopathy pathophysiology
- Restrictive cardiomyopathy signs and symptoms
- Restrictive cardiomyopathy complications
- Restrictive cardiomyopathy diagnosis
- Restrictive cardiomyopathy differential diagnosis
- Restrictive cardiomyopathy treatment
- Restrictive cardiomyopathy prognosis
- Restrictive cardiomyopathy lifespan
Restrictive cardiomyopathy
Restrictive cardiomyopathy (RCM) also called infiltrative cardiomyopathy or constrictive cardiomyopathy, is a rare group of heart disease that occurs when your heart ventricles (lower chambers) muscle becomes stiff, looses its flexibility and can’t fill with blood 1, 2. Restrictive cardiomyopathy (RCM) is characterized by severely enlarged atria (atrial dilatation), normal-sized ventricles (non-dilated ventricles) with increased myocardial stiffness leading to impaired ventricular filling and diastolic dysfunction but with normal systolic function and normal ventricular wall thicknesses 1, 3, 2. Systolic function at least in the beginning of the disease is near normal but might be reduced at later stages of the disease 4. Sometimes also a mild hypertrophy is observed, making diagnostic distinction between restrictive cardiomyopathy and hypertrophic cardiomyopathy difficult 5. Restrictive cardiomyopathy (RCM) prevents your heart from filling with blood, which can lead to heart failure that increases pressure on your heart and may cause fluid buildup in your lungs (pulmonary edema). Restrictive cardiomyopathy (RCM) patients present with symptoms of left and/or right ventricular heart failure with preserved ejection fraction (HFpEF), atrial fibrillation, ventricular arrhythmias and frequently conduction disorders 6. Symptoms of restrictive cardiomyopathy (RCM) vary depending on the severity. Non-specific restrictive cardiomyopathy treatment options include fluid and salt (sodium) restrictions and medical treatment of heart failure with reduction of volume overload as well as anticoagulation and antiarrhythmic therapy. Very often heart transplantation is the only option for long-term survival 7.
Restrictive cardiomyopathy is a fairly rare disease, but it accounts for approximately 5% of all cases of cardiomyopathy 2. Most causes of restrictive cardiomyopathy are acquired 8.
There are multiple restrictive cardiomyopathy types that exist and vary in their cause, clinical presentation, diagnostic evaluation, treatment, and prognosis. Three of the leading causes of restrictive cardiomyopathy (RCM) are cardiac amyloidosis, cardiac sarcoidosis, and cardiac hemochromatosis 2, 9:
- Cardiac amyloidosis is a condition where abnormal proteins build up in your heart muscle making it difficult for your heart to pump blood 10. You can inherit cardiac amyloidosis or it can develop on its own (usually later in life). As abnormal proteins accumulate in your heart muscle, your heart struggles to pump, ultimately leading to heart failure and death. However, cardiac amyloidosis is usually treatable and in some cases is curable.
- Cardiac sarcoidosis is a rare autoimmune disease where tiny collections of immune cells form granulomas (a small cluster of immune cells) in your heart muscle 9, 11. Cardiac sarcoidosis occurs when immune cells clump together to form granulomas in your heart tissue. This can result in heart rhythm abnormalities (arrhythmias), such as ventricular tachycardia or heart block. It can also lead to cardiomyopathy or heart failure. Cardiac sarcoidosis is treated with immunosuppressant drugs such as steroids and if you have arrhythmias or heart failure, heart failure medications. Additionally, some patients may require heart procedures such as implantable cardioverter defibrillator (ICD), pacemaker, catheter ablation and heart transplant or left ventricular assist device.
- Cardiac hemochromatosis also called primary iron-overload cardiomyopathy is a condition where iron builds up in your heart muscle causing arrhythmias and heart failure 12, 13, 14. Cardiac hemochromatosis causes a dilated cardiomyopathy with dilated ventricles, low left ventricular ejection fraction (LVEF), and decreased fractional shortening 15, 16. Cardiac hemochromatosis is a potentially preventable cause of heart failure. Unfortunately, cardiac hemochromatosis is often diagnosed in the later stages of the disease, when complications may have already developed. Cardiac hemochromatosis is treated with therapeutic phlebotomy (drawing blood from a vein) to remove iron from your body 17. Additional treatment include iron chelation to reduce iron levels and lifestyle changes such as avoiding iron supplements, vitamin C supplements, alcohol, and raw fish and shellfish.
Patients with restrictive cardiomyopathy primarily present with advanced disease and obvious signs of cardiopulmonary compromise, but in some circumstances, the diagnosis is incidental 2. It is important to suspect restrictive cardiomyopathy in any patient with a normal or close to normal systolic function and evidence of diastolic dysfunction with a restrictive filling pattern on echocardiogram 2. For those that present symptomatically, there is a wide range of presentations. Some may present in full-blown heart failure (jugular venous distension, ascites, lower extremity edema, and less commonly pulmonary edema). Some may complain of poor exercise tolerance or be newly diagnosed with an arrhythmia such as atrial fibrillation. In the less fortunate cases, some present as sudden cardiac arrest. Other less common presentations include ischemia, thrombus, and misdiagnosed as hypertrophic cardiomyopathy with left ventricular outflow obstruction. When performing the evaluation and physical in a patient suspected of restrictive cardiomyopathy, it is essential to look for extracardiac manifestations such as carpal tunnel, which may be present in amyloidosis or bilateral hilar infiltrates seen in sarcoidosis 18. Hemochromatosis may present with the classic bronze skin, cirrhosis, arthralgias, and endocrinopathies such as diabetes mellitus 19.
The cardiac impulse is less displaced than in dilated cardiomyopathy and less dynamic than in hypertrophic cardiomyopathy. A fourth heart sound (S4) is more common than a third heart sound (S3) in sinus rhythm, but atrial fibrillation (AF) is common. Jugular venous pressures often show rapid Y descents and may increase during inspiration (positive Kussmaul’s sign). Kussmaul’s sign is increased jugular venous pressure (JVP) with inhalation instead of decreasing.
Restrictive cardiomyopathy is a complex diagnosis that is best that your primary care doctor refer your to a cardiologist. Restrictive cardiomyopathy can be difficult to diagnose and requires thorough testing, which may include the following:
- Electrocardiogram (ECG or EKG). Electrocardiogram (ECG) measures how electricity is functioning in your heart. People may also refer to it as an electrocardiography. Electrocardiogram (ECG) is one of the first tools used when a cardiac diagnosis is suspected. For example, a low-to-normal voltage in the QRS complex despite thickened cardiac muscle in the absence of valvular or hypertensive disease may lead one to suspect amyloidosis related restrictive cardiomyopathy. However, the absence of low voltage does not exclude the diagnosis 20.
- Echocardiogram. An echocardiogram is an ultrasound test that checks the structure and function of your heart. An echo can diagnose a range of conditions including cardiomyopathy and valve disease. The echocardiogram is the primary radiographic diagnostic test for identifying patients with restrictive cardiomyopathy. It may help aid in differentiating restrictive cardiomyopathy from some of its common imitators, such as constrictive pericarditis. The echocardiogram may also provide information to suggest a specific diagnosis.
- Cardiac MRI. Cardiac magnetic resonance (CMR) imaging can also aid in the diagnosis. Cardiac MRI can be useful to gain ancillary evidence to support a diagnosis of restrictive cardiomyopathy, specifically in amyloidosis, where a gadolinium enhancement pattern is highly suggestive of amyloid.
- Cardiac PET scan. Positron emission tomography (PET) scan is a painless imaging test that uses radioactive substances to show how organs and tissues are functioning. PET scans are mainly used to assess cancers, neurological (brain) diseases and cardiovascular (heart-related) disease. PET scan can also be used to aid in diagnosis as it also demonstrates an affinity for amyloid 21.
- Right heart catheterization also known as a pulmonary artery catheterization. A right heart catheterization is a procedure that involves inserting a thin tube (catheter) into the right side of your heart. Right heart catheterization is used to assess your heart’s function, to see how well your heart is pumping, and and to measure the pressures in your heart and lungs
- Cardiac biopsy. Endomyocardial biopsy (EMB) may help establish the diagnosis in some cases if the primary workup has not been able to do so. Endomyocardial biopsy is the gold standard for the diagnosis of cardiac amyloidosis 20. Fat pad aspiration is positive in about 50% of cases. Mass spectrometry, the gold standard for typing the tissue, is done following biopsy to evaluate further the disease process. Mutation analysis can diagnose hereditary hemochromatosis, especially due to the strong association with the HFE C282Y and H63D.
Treatment for restrictive cardiomyopathy includes treating the underlying cause and heart failure symptoms that may arise secondary to the disease. Currently, there is no cure for restrictive cardiomyopathy, but there are some treatments available to alleviate the symptoms of the disease 2. For heart failure symptoms, diuretics (water pills) are the mainstay of treatment to reduce volume overload but must be monitored closely to prevent excessive diuresis as patients with restrictive cardiomyopathy rely on high filling pressures to maintain cardiac output 2. The use of beta-blockers or calcium channel blockers is sometimes introduced to increase the filling time 22. They may also be beneficial in treating arrhythmias, which are common in this patient population. Angiotensin-receptor blockers may also be used, especially if concurrent systolic heart failure develops.
For sarcoidosis, antiarrhythmics are a common therapy choice due to the high incidence of conduction disease 2. Immunosuppressive agents such as corticosteroids and steroid-sparing agents are also sometimes used to treat sarcoidosis 2. For hemochromatosis, the treatment of choice is therapeutic phlebotomy. Advanced heart failure treatment, such as cardiac transplant or left ventricular assist devices, may be appropriate for some patients 21. Ultimately, the choice of a specific therapy depends on the clinical condition, the risk of dangerous events, and the ability of the patient to tolerate the therapy 2.
Similar to the other cardiomyopathies, restrictive cardiomyopathy (RCM) has a very poor prognosis, having the worst prognosis as compared to all other cardiomyopathies 23, 2. Statistical studies report only a 2 to 5 year survival rate in patients with restrictive cardiomyopathy (RCM) 21.
Figure 1. Human heart anatomy
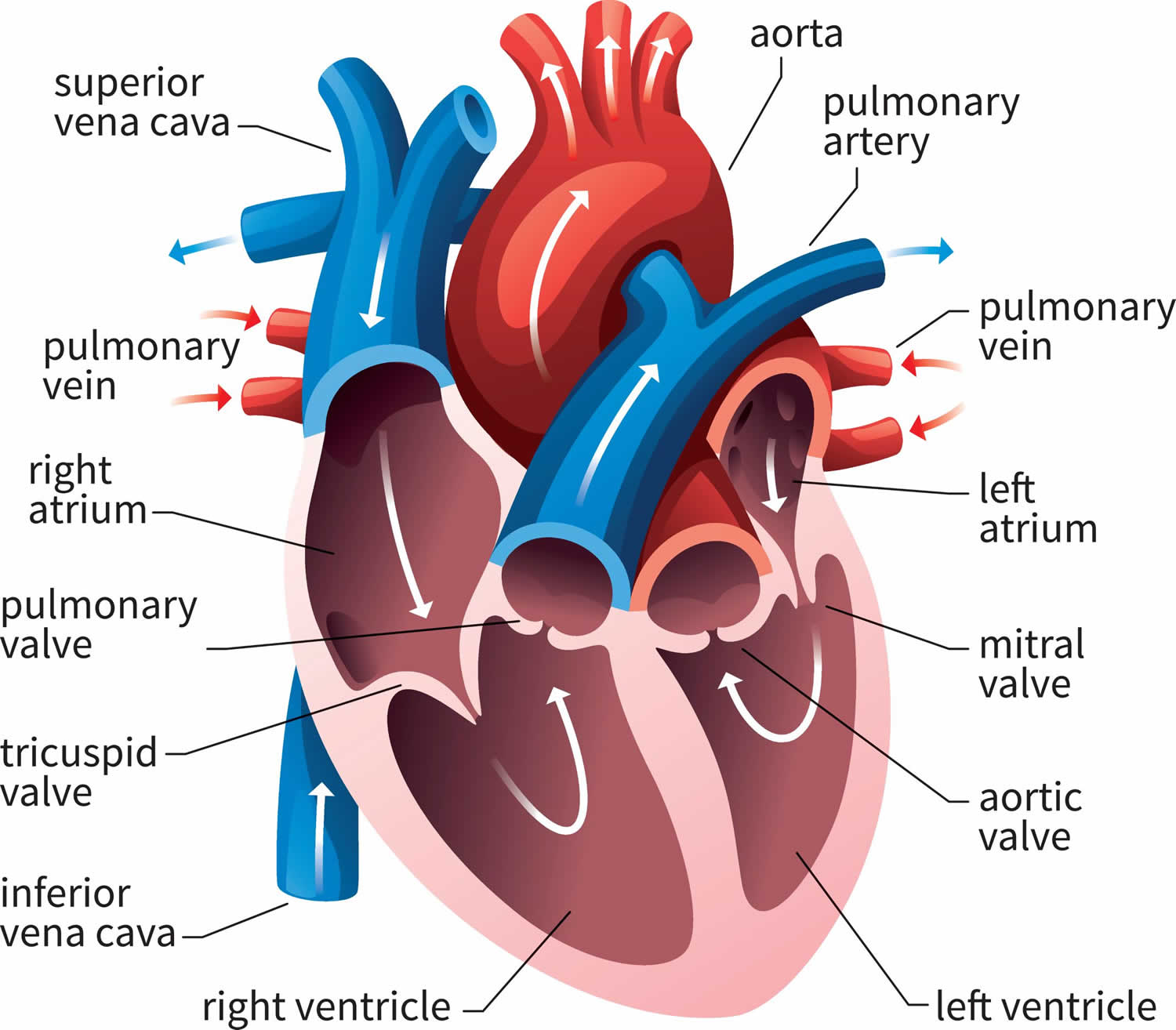
Figure 2. Restrictive cardiomyopathy
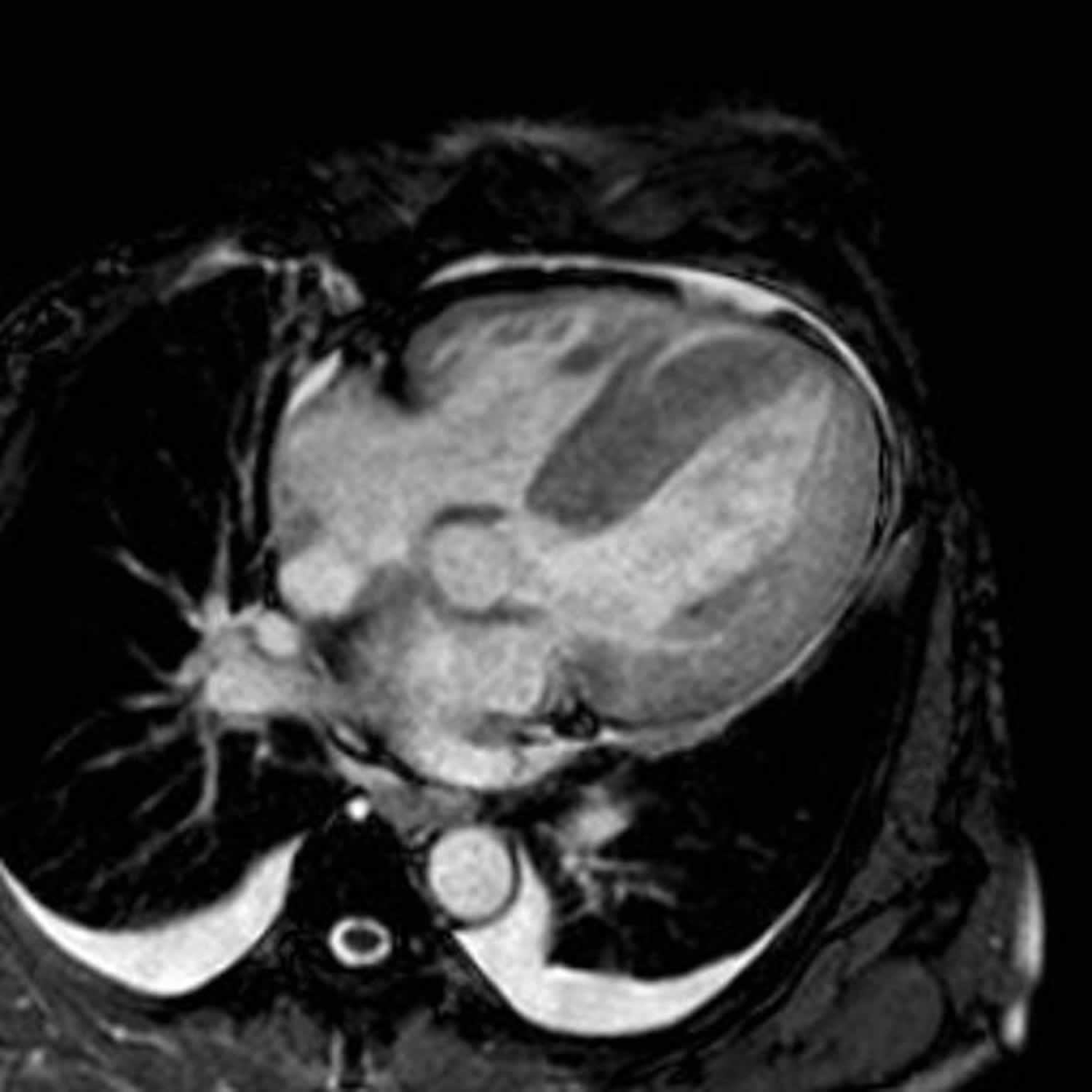
Footnotes: 55 year old female was on treatment for multiple myeloma, when she started to suffer from progressive shortness of breath (dyspnea). Her heart MRI scans showed all four heart chambers have thickened walls, including the atria and atrial septum, which alone is very suggestive of amyloidosis, even without other findings. The hallmarks of restrictive cardiomyopathy are thickened cardiac walls (due to infiltration) and restricted diastolic filling of the ventricles with lowered ejection fraction (EF) as opposed to unaltered or even increased EF with hypertrophic cardiomyopathy. However, usually at least some late enhancement at cardiac amyloidosis can be seen, and this patient was no exception. Late enhancement was seen ranging from smaller sub-endocardial and mid-myocardial patches to a transmural larger patch of late enhancement in the basal part of the heart. Additional findigs are pericardial and pleural effusions. This patient had exclusively cardiac AL amyloidosis and underwent further treatment with stem cell transplantation. Eight months after this cardiac MR was performed, she is still alive and feeling quite well.
[Source 24 ]Figure 3. Restrictive cardiomyopathy echocardiography
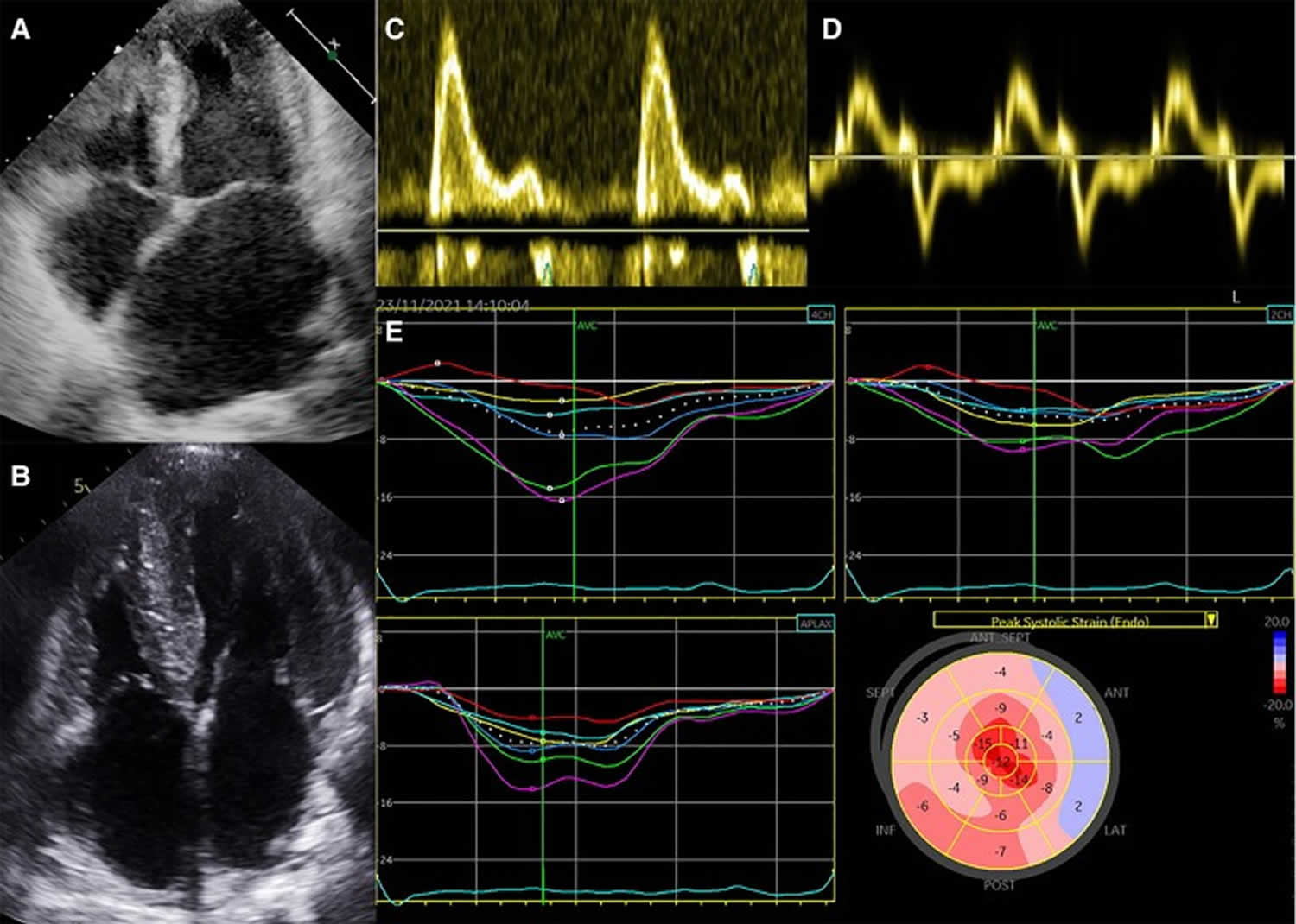
Footnotes: Echocardiography in restrictive cardiomyopathies. (A) Small left ventricular cavity size in presence of significantly increased wall thickness and severe left atrial dilatation; (B) biventricular wall thickening in absence of pulmonary hypertension; (C and D) Restrictive filling pattern with elevated E/E′ ratio in keeping with increased left ventricular filling pressures; (E) myocardial strain analysis showing an apical sparing pattern in a patient with cardiac amyloidosis.
[Source 1 ]Figure 4. Restrictive cardiomyopathy diagnostic algorithm
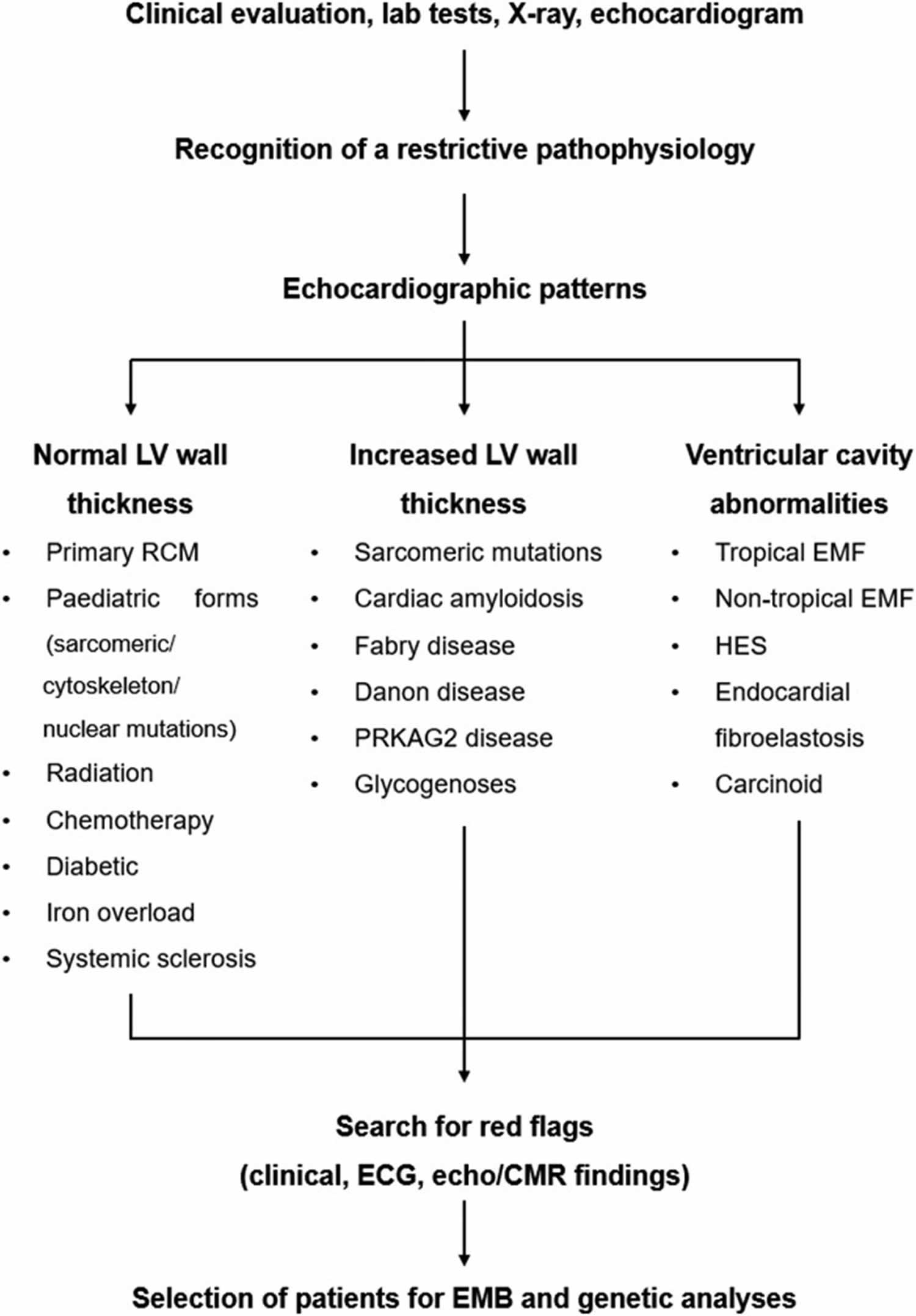
Footnotes: Red flag-based diagnostic approach. The main disorders presenting with normal or increased left ventricular wall thickness are listed.
Abbreviations: CMR = cardiovascular magnetic resonance; EMB = endomyocardial biopsy; EMF = endomyocardial fibrosis; HES = hypereosinophilic syndrome; LV = left ventricle; PRKAG2 = protein kinase AMP-activated non-catalytic subunit gamma-2; RCM = restrictive cardiomyopathy.
[Source 1 ]How does restrictive cardiomyopathy affect me?
Your heart has four chambers made of muscle that contract to help pump your blood. The two upper chambers are atria, and the two lower chambers are ventricles (see Figure 1 above). Restrictive cardiomyopathy causes the ventricles to become rigid and unable to fill with blood completely. Restrictive cardiomyopathy affects how your heart function including how blood flows through your heart. Restrictive cardiomyopathy patients can present with symptoms and signs of left ventricular failure and/or right ventricular failure 25.
Who does restrictive cardiomyopathy affect?
Restrictive cardiomyopathy can affect anyone. But the underlying conditions that cause restrictive cardiomyopathy are more common in some people than others. For example, sarcoidosis, which causes restrictive cardiomyopathy, is most prevalent in females who are Black.
How common is restrictive cardiomyopathy?
Because of the heterogenous nature of the origins and manifestations of the restrictive cardiomyopathies and the concomitant challenges in diagnosing these diseases, it is difficult to accurately estimate the incidence and prevalence of any of the restrictive cardiomyopathies 26, 21. However, cardiomyopathy is a common heart muscle disease. Up to 1 in 500 people may have cardiomyopathy 27. But restrictive cardiomyopathy is the rarest type of cardiomyopathy — about 5% of all cardiomyopathies are restrictive cardiomyopathy. There are also regional differences among the prevalence of restrictive cardiomyopathy according to the cause 21. For example, endomyocardial fibrosis is primarily seen in the tropics and sub-Saharan Africa 28, whereas cardiac amyloidosis is more commonly diagnosed in other regions. Most causes of restrictive cardiomyopathy are acquired. However, mapping several gene mutations as a cause of restrictive cardiomyopathy have been recognized. These include mutations in the sarcomere subunits, such as troponin T (TNNT2 gene), troponin I (TNNI3), α-actin (ACTC), and β-myosin heavy chain (MYH7). Most of these mutations are inherited in an autosomal dominant fashion 29.
How can I reduce my risk of restrictive cardiomyopathy?
Restrictive cardiomyopathy makes your heart work harder to pump blood. Talk to your heart specialist (cardiologist) about steps you can take to reduce your heart’s workload.
While you can’t prevent the underlying conditions that cause restrictive cardiomyopathy, you can help keep your heart healthy by:
- Controlling your blood pressure.
- Maintaining a healthy weight and diet.
- Reducing your stress.
- Exercise regularly.
- Don’t smoke or quit smoking.
Restrictive cardiomyopathy causes
There are many causes of restrictive cardiomyopathy (RCM) that include infiltrative diseases, storage diseases, and a variety of systemic diseases. Infiltrative diseases are pathologies that lead to a build-up of a substance in the heart muscle (myocardium). These diseases include amyloidosis (AL, ATTRm, ATTRwt, ApoA-I), sarcoidosis, hereditary hemochromatosis, as well as primary hyperoxaluria 9. Amyloidosis is the most common infiltrative disease. Storage diseases are rare inherited abnormalities in metabolism. Storage diseases include Fabry disease, Gaucher disease, glycogen storage disease (a rare genetic disorder that prevents the body from storing or breaking down glycogen [a form of sugar that the body uses for energy]), mucopolysaccharidosis type 2 (Hunter syndrome), Niemann-Pick disease, Danon disease, and Friedrich ataxia. Several different systemic diseases can also lead to restrictive cardiomyopathy. Some of these pathologies include diabetes, scleroderma, myofibrillar myopathies, pseudoxanthoma elasticum, Werner syndrome, sarcomeric protein disorders (genetic or acquired conditions that affect the proteins in the sarcomere, the smallest contractile unit of muscle), carcinoid cardiomyopathy (a rare condition that is caused by excess serotonin secretion from a tumor [carcinoid syndrome] which causes plaque and scar tissue to build up in the heart valves and muscle), idiopathic fibrosis, hypereosinophilic fibrosis, chronic eosinophilic leukemia fibrosis, endocardial fibroelastosis, and metastatic cancers 2. Other causes of restrictive cardiomyopathy include idiopathic causes, including radiation therapy and some medications 30. Some drugs known for causing restrictive cardiomyopathy include anthracycline, busulfan, ergotamines, methysergide, mercurial agents, and serotonin-containing agents 21, 22.
You may develop restrictive cardiomyopathy if you have:
- Amyloidosis: Amyloidosis is a rare disease that occurs when abnormal protein called amyloid builds up in your organs. The buildup of these proteins called amyloid fibrils can make your organs not work properly and lead to organ failure. Organs that may be affected include the heart, kidneys, liver, spleen, nervous system and digestive tract. Amyloidosis is the most common cause of restrictive cardiomyopathy in the United States. Amyloidosis affects men and women equally. AL amyloidosis (amyloid light chain or primary amyloidosis) is the most common cause of restrictive cardiomyopathy. The wild-type transthyretin amyloidosis is most often found in the elderly population. The V1221 mutant transthyretin has a higher rate of symptomatic heart failure 21, 10.
- Sarcoidosis: Sarcoidosis is a rare disease that causes tiny collections of immune system cells (granulomas) to form in your body’s organs. Sarcoidosis can affect your lungs, skin, lymph nodes, eyes, heart, and nervous system. Sarcoidosis is more common in women than men. The highest prevalence of sarcoidosis is among black women. The highest incidence worldwide of sarcoidosis is in Japan 9, 11.
- Hemochromatosis: Hemochromatosis is a genetic disorder that causes your body to store too much iron, which can damage organs. Without treatment, hemochromatosis can cause iron overload, a buildup of iron that can damage many parts of your body, including your liver, heart, pancreas, endocrine glands, and joints. Hemochromatosis affects men and women equally. Hemochromatosis prevalence is 1 per 200 individuals. Men are more likely to have cirrhosis (liver scarring) associated with hemochromatosis. The predominant gene mutation is HFE C282Y. It is inherited in an autosomal recessive pattern. Another common mutation seen in hemochromatosis is the H63D 14.
- Loffler endocarditis also known as eosinophilic endomyocardial disease or fibroplastic endocarditis: Loeffler endocarditis is a rare heart disease that causes the heart muscle to stiffen and malfunction 31. The predominant pathology of Loeffler endocarditis is diffuse eosinophilic infiltration of the heart muscle (myocardium) 31. First described by W. Loeffler in 1936, Loeffler endocarditis is associated with peripheral eosinophilia and is one of the rare complications of hypereosinophilic syndromes 31. Extensive eosinophilic infiltration and damage to multiple organs characterize hypereosinophilic syndromes 32. Loeffler endocarditis results in impaired heart relaxation with impaired diastolic filling 33. Loeffler endocarditis is common in tropical climates and sub-Saharan Africa. Up to twenty percent of heart failure in this region is thought to be secondary to endomyocardial fibrosis 34. Cardinal manifestations of Loeffler endocarditis include left or right heart failure, thromboembolic events (stroke, limb ischemia, renal infarction), or arrhythmia. Life-threatening clinical presentations warrant immediate initiation of therapy, including steroids or immunosuppressive therapy 35, 36. Endomyocardial fibrosis is a disease closely resembling the late stage of Loeffler endocarditis.
- Connective tissue disease. Connective tissue disease is an umbrella term for a group of disorders that affect the body’s connective tissues. Connective tissues are made of collagen and elastin, that connect and support your organs and body structure. Connective tissues hold your body’s structures together. There are over 200 known connective tissue disorders. They fall into three main categories:
- Autoimmune diseases. Autoimmune diseases are what many people think of when they think of connective tissue disease. In these conditions, your immune system generates chronic inflammation in some parts of your body. Chronic inflammation causes pain, swelling and, eventually, permanent damage to your tissues.
- Lupus also known as systemic lupus erythematosus (SLE): Systemic lupus erythematosus (SLE) can cause inflammation in any of your connective tissues, and sometimes all of them. Systemic lupus erythematosus (SLE) may affect your skin, joints and vital organs, like your heart, lungs and kidneys.
- Scleroderma: Scleroderma is a rare autoimmune disease that causes your body to overproduce collagen, which can cause thickening and hardening of your skin and organs, including your digestive system, kidneys, heart and lungs. Scleroderma can also affect the muscles, bones, blood vessels, and internal organs. Scleroderma affects many more women than men, and it’s typically found in people between the ages of 30 and 50.
- Genetic disorders. Genetic disorders of the connective tissue result from a gene mutation that you inherit at birth. The mutation affects how your connective tissues develop. It usually affects one of the two primary building blocks in all connective tissues: collagen or elastin. This causes various defects in your tissues.
- Cancers (sarcomas). The type of cancer that can start in your connective tissues is called sarcoma. Sarcomas can start in your bones, cartilage, fat, muscles, ligaments, tendons or the deep layers of your skin. They can also start in other “soft tissues” that aren’t technically connective tissues, like epithelium and endothelium.
- Autoimmune diseases. Autoimmune diseases are what many people think of when they think of connective tissue disease. In these conditions, your immune system generates chronic inflammation in some parts of your body. Chronic inflammation causes pain, swelling and, eventually, permanent damage to your tissues.
You’re also more likely to develop restrictive cardiomyopathy after certain cancer treatments, including:
- Chemotherapy.
- Radiation therapy also known as radiation cardiotoxicity or radiation-induced heart disease.
You’re also likely to develop restrictive cardiomyopathy after use of certain drugs 37, 38:
- Hydroxychloroquine (used to treat SLE and other connective tissue disorders)
- Methysergide
- Anthracyclines.
Restrictive cardiomyopathy pathophysiology
Restrictive cardiomyopathy may result from inherited or acquired disorders and disease or a combination thereof, which broadly can be classified as infiltrative, storage disease, noninfiltrative, and endomyocardial 21. Most restrictive cardiomyopathies are due to infiltration of abnormal substances between heart muscle cells (myocytes), storage of abnormal metabolic products within heart muscle cells or fibrotic injury 21. Restrictive cardiomyopathy is predominantly a disease of diastolic dysfunction where the systolic (contractile) of the myocardium is usually unaffected.
Infiltrative (between heart muscle cells)
- Amyloidosis
- AL amyloidosis (amyloid light chain or primary amyloidosis)
- Familial (abnormal transthyretin)
- Senile (normal transthyretin or atrial peptides)
- Primary hyperoxaluria
- Inherited metabolic defects
Storage (within heart muscle cells)
- Hemochromatosis (iron overload)
- Inherited metabolic defects
- Fabry disease
- Gaucher disease
- Glycogen storage disease (2 and 3)
- Mucopolysaccharidosis type 1 (Hurler syndrome)
- Mucopolysaccharidosis type 2 (Hunter syndrome)
- Niemann–Pick disease
Noninfiltrative cardiomyopathy
- Idiopathic
- Diabetic cardiomyopathy
- Scleroderma
- Myofibrillar myopathies
- Pseuxanthoma elasticum
- Sarcomeric protein disorders
- Werner’s syndrome.
Endomyocardial fibrosis
- Endomyocardial fibrosis
- Endocardial fibroelastosis
- Tropical endomyocardial fibrosis
- Hypereosinophilic syndrome (Loeffler endocarditis)
- Chronic eosinophilic leukemia
- Carcinoid heart disease
- Consequence of cancer/cancer therapy
- Radiation
- Drugs (anthracyclines)
- Metastatic cancer
- Drugs: e.g., serotonin, methysergide, ergotamine, mercurial agents, busulfan.
Restrictive cardiomyopathy signs and symptoms
You may not have any symptoms of restrictive cardiomyopathy. But as the condition worsens, you might develop heart failure symptoms. These may include:
- Chest pain (at rest or with exercise).
- Dizziness or fainting.
- Fatigue.
- Heart palpitations.
- Shortness of breath (dyspnea).
- Weight gain.
- Edema in your feet and legs.
- Bloating or nausea.
Restrictive cardiomyopathy complications
Restrictive cardiomyopathy complications may include the following 21, 39:
- Thromboembolism
- Arrhythmias
- Heart failure
- Cardiac cirrhosis also known as congestive hepatopathy, a condition where the liver is damaged due to right-sided heart failure.
- Other complications depend on the underlying cause of restrictive cardiomyopathy.
Restrictive cardiomyopathy diagnosis
Restrictive cardiomyopathy is a complex diagnosis that is best that your primary care doctor refer your to a cardiologist. Restrictive cardiomyopathy can be difficult to diagnose and requires thorough testing, which may include the following:
- Electrocardiogram (ECG or EKG). Electrocardiogram (ECG) measures how electricity is functioning in your heart. People may also refer to it as an electrocardiography. Electrocardiogram (ECG) is one of the first tools used when a cardiac diagnosis is suspected. For example, a low-to-normal voltage in the QRS complex despite thickened cardiac muscle in the absence of valvular or hypertensive disease may lead one to suspect amyloidosis related restrictive cardiomyopathy. However, the absence of low voltage does not exclude the diagnosis 20.
- Echocardiogram. An echocardiogram is an ultrasound test that checks the structure and function of your heart. An echo can diagnose a range of conditions including cardiomyopathy and valve disease. The echocardiogram is the primary radiographic diagnostic test for identifying patients with restrictive cardiomyopathy. It may help aid in differentiating restrictive cardiomyopathy from some of its common imitators, such as constrictive pericarditis. The echocardiogram may also provide information to suggest a specific diagnosis.
- The first clue of restrictive pathophysiology is the combination of biatrial enlargement (which cannot be attributed to specific causes such as valve disease or atrial fibrillation [AF]), normal or mildly reduced left ventricle (LV) and right ventricle (RV) ejection fraction and non-dilated ventricles. Doppler imaging can then show a restrictive filling pattern of transmitral flow with increased early diastolic filling velocity (E wave) due to elevated left atrium (LA) pressure, and decreased atrial filling velocity (A wave) due to the high ventricular diastolic pressure, reduction of mitral deceleration time, and isovolumetric relaxation time. Additionally, the ratio between systolic and diastolic pulmonary venous flow ratios is markedly reduced because of high left atrium (LA) pressures. Tissue Doppler typically shows reduced early diastolic myocardial velocity (e′) leading to an elevated E/e′ ratio. Congestion of the inferior vena cava and hepatic veins and diastolic flow reversal in the hepatic veins during inspiration are common, following the inability of a non-compliant right ventricle (RV) to accommodate the increased venous return 40, 41, 42.
- Cardiac MRI. Cardiac magnetic resonance (CMR) imaging can also aid in the diagnosis. Cardiac MRI can be useful to gain ancillary evidence to support a diagnosis of restrictive cardiomyopathy, specifically in amyloidosis, where a gadolinium enhancement pattern is highly suggestive of amyloid.
- Whenever feasible, cardiac magnetic resonance (CMR) should be part of the diagnostic work-up of patients with suspected restrictive cardiomyopathy. Cardiovascular magnetic resonance represents the gold standard non-invasive technique to quantify biventricular volumes, mass, and EF, with cine steady-state free-precession sequences. Furthermore, cardiac magnetic resonance (CMR) allows to characterize myocardial tissue properties: myocardial oedema is typically detected by T2-weighted imaging, intraventricular thrombosis with early gadolinium enhancement, and myocardial interstitial expansion with late gadolinium enhancement (LGE), usually due to fibrosis or amyloid extracellular deposition, sometimes also to myocyte necrosis or extracellular edema. Native (i.e. pre-contrast) T1- and T2-mapping sequences provide a quantitative assessment of myocardial tissue changes; after gadolinium injection, myocardial perfusion mapping and extracellular volume (ECV) mapping provide a quantitative assessment of myocardial perfusion and of the extracellular space, respectively 43, 44, 45.
- Cardiac PET scan. Positron emission tomography (PET) scan is a painless imaging test that uses radioactive substances to show how organs and tissues are functioning. PET scans are mainly used to assess cancers, neurological (brain) diseases and cardiovascular (heart-related) disease. PET scan can also be used to aid in diagnosis as it also demonstrates an affinity for amyloid 21.
- Right heart catheterization also known as a pulmonary artery catheterization. A right heart catheterization is a procedure that involves inserting a thin tube (catheter) into the right side of your heart. Right heart catheterization is used to assess your heart’s function, to see how well your heart is pumping, and and to measure the pressures in your heart and lungs
- Cardiac biopsy. Endomyocardial biopsy (EMB) may help establish the diagnosis in some cases if the primary workup has not been able to do so. Endomyocardial biopsy is the gold standard for the diagnosis of cardiac amyloidosis 20. Fat pad aspiration is positive in about 50% of cases. Mass spectrometry, the gold standard for typing the tissue, is done following biopsy to evaluate further the disease process. Mutation analysis can diagnose hereditary hemochromatosis, especially due to the strong association with the HFE C282Y and H63D.
Histopathology
The histology of restrictive cardiomyopathies varies depending on cause. It is specifically useful with infiltrative cardiomyopathies and some of the storage diseases. A list of common histology associations are listed below 46, 47, 48, 49, 50, 51, 52:
- Amyloidosis: positive Congo red stain
- Sarcoidosis: granulomatosis reaction
- Primary hyperoxaluria: oxalate deposition
- Niemann-Pick disease: foam cells
- Mucopolysaccharidosis type 2: glycosaminoglycans
- Glycogen storage disease: dicarboxylic acids
- Hereditary hemochromatosis (iron)
- Gaucher disease: Gaucher cells (lipid-laden macrophages)
- Fabry disease: ceramide trihexoside
Restrictive cardiomyopathy differential diagnosis
Restrictive cardiomyopathy differential diagnosis may include the following 21:
- Constrictive pericarditis
- Acute or chronic heart failure
- Hypertensive heart disease
- Hypertrophic cardiomyopathy
- Acute or chronic pericarditis
Constrictive pericarditis is the most commonly mistaken for restrictive cardiomyopathy (RCM) out of the list above. That is why it is essential to explore the similarities and differences in more detail. The two diseases present almost identically, except for a few key signs and symptoms 2. Jugular venous pressure (JVP) elevation or distension, Kussmaul sign (a condition where the jugular venous pressure [JVP] increases instead of decreasing when a person inhales), and diastolic sounds are both seen in restrictive cardiomyopathy and constrictive pericarditis 2. However, there are some subtle differences. For example, third heart sound (S3) and elevated B-type natriuretic peptide (BNP) are far more common in restrictive cardiomyopathy 2. Whereas a pericardial knock, pericardial calcifications on chest x-ray, pericardial thickening on imaging, and BNP levels less than 100 are more likely seen in constrictive pericarditis 2. Furthermore, one clear difference between the constrictive pericarditis and restrictive cardiomyopathy (RCM) is the presence of ventricular interdependence. Ventricular dependence seen only in constrictive pericarditis is described as an increased filling of one of the ventricles only, with a reciprocal decreased filling of the other ventricle 40.
Restrictive cardiomyopathy treatment
There’s no specific treatment for restrictive cardiomyopathy. Your heart speacialist (cardiologist) will treat the underlying cause of your condition. If you have heart failure symptoms, your cardiologist may treat you with:
- Corticosteroids (if you have sarcoidosis).
- Diuretics (water pills).
- Medications to treat heartbeat irregularities (antiarrhythmics, beta blockers or calcium channel blockers).
- Therapeutic phlebotomy (blood removal for hemochromatosis).
- Medications to treat certain types of amyloidosis.
Some people with restrictive cardiomyopathy may ultimately need heart transplant surgery. Your cardiologist may also recommend palliative care for this condition.
Restrictive cardiomyopathy prognosis
Similar to the other cardiomyopathies, restrictive cardiomyopathy (RCM) has a very poor prognosis, having the worst prognosis as compared to all other cardiomyopathies 23, 2. Statistical studies report only a 2 to 5 year survival rate in patients with restrictive cardiomyopathy (RCM) 21.
Restrictive cardiomyopathy lifespan
Restrictive cardiomyopathy occurs when muscles in your heart’s ventricles become rigid. The ventricles can’t fill with blood. Your heart must work too hard to pump blood, causing heart failure. The survival rate for people with restrictive cardiomyopathy varies. Your cardiologist can help you and your family know what to expect as your condition progresses. Statistical studies report only a 2 to 5 year survival rate in patients with restrictive cardiomyopathy (RCM) 21.
- Rapezzi C, Aimo A, Barison A, Emdin M, Porcari A, Linhart A, Keren A, Merlo M, Sinagra G. Restrictive cardiomyopathy: definition and diagnosis. Eur Heart J. 2022 Dec 1;43(45):4679-4693. doi: 10.1093/eurheartj/ehac543[↩][↩][↩][↩]
- Brown KN, Pendela VS, Ahmed I, et al. Restrictive Cardiomyopathy. [Updated 2023 Jul 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537234[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Brodehl A, Gerull B. Genetic Insights into Primary Restrictive Cardiomyopathy. J Clin Med. 2022 Apr 8;11(8):2094. doi: 10.3390/jcm11082094[↩]
- Cimiotti D, Budde H, Hassoun R, Jaquet K. Genetic Restrictive Cardiomyopathy: Causes and Consequences-An Integrative Approach. Int J Mol Sci. 2021 Jan 8;22(2):558. doi: 10.3390/ijms22020558[↩]
- Tariq M. Importance of Genetic Evaluation and Testing in Pediatric Cardiomyopathy. World J. Cardiol. 2014;6:1156. doi: 10.4330/wjc.v6.i11.1156[↩]
- Seferovic P.M., Polovina M., Bauersachs J., Arad M., Gal T.B., Lund L.H., Felix S.B., Arbustini E., Caforio A.L.P., Farmakis D., et al. Heart failure in cardiomyopathies: A position paper from the heart failure association of the european society of cardiology. Eur. J. Heart Fail. 2019;21:553–576. doi: 10.1002/ejhf.1461[↩]
- DePasquale E.C., Nasir K., Jacoby D.L. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J. Heart Lung Transplant. 2012;31:1269–1275. doi: 10.1016/j.healun.2012.09.018[↩]
- Rapezzi C, Ortolani P, Traini AM, Caporale R, Ferlito M, Branzi A, Magnani B. Le cardiomiopatie restrittive [Restrictive cardiomyopathies]. Cardiologia. 1993 Dec;38(12 Suppl 1):283-8. Italian.[↩]
- Costabel U, Wessendorf TE, Bonella F. Epidemiologie und klinisches Erscheinungsbild der Sarkoidose [Epidemiology and Clinical Presentation of Sarcoidosis]. Klin Monbl Augenheilkd. 2017 Jun;234(6):790-795. German. doi: 10.1055/s-0042-105569[↩][↩][↩][↩]
- Kyriakou P, Mouselimis D, Tsarouchas A, Rigopoulos A, Bakogiannis C, Noutsias M, Vassilikos V. Diagnosis of cardiac amyloidosis: a systematic review on the role of imaging and biomarkers. BMC Cardiovasc Disord. 2018 Dec 4;18(1):221. doi: 10.1186/s12872-018-0952-8[↩][↩]
- Nunes H, Freynet O, Naggara N, Soussan M, Weinman P, Diebold B, Brillet PY, Valeyre D. Cardiac sarcoidosis. Semin Respir Crit Care Med. 2010 Aug;31(4):428-41. doi: 10.1055/s-0030-1262211[↩][↩]
- Aronow WS. Management of cardiac hemochromatosis. Arch Med Sci. 2018 Apr;14(3):560-568. doi: 10.5114/aoms.2017.68729[↩]
- Kremastinos DT, Farmakis D. Iron overload cardiomyopathy in clinical practice. Circulation. 2011 Nov 15;124(20):2253-63. doi: 10.1161/CIRCULATIONAHA.111.050773[↩]
- Loréal O, Cavey T, Robin F, Kenawi M, Guggenbuhl P, Brissot P. Iron as a Therapeutic Target in HFE-Related Hemochromatosis: Usual and Novel Aspects. Pharmaceuticals (Basel). 2018 Nov 26;11(4):131. doi: 10.3390/ph11040131[↩][↩]
- Skinner C, Kenmure AC. Haemochromatosis presenting as congestive cardiomyopathy and responding to venesection. Br Heart J. 1973;35:466–8. doi: 10.1136/hrt.35.4.466[↩]
- Cascales A, Sanchez-Vega B, Navarro N, et al. Clinical and genetic determinants of anthracycline-induced cardiac iron accumulation. Int J Cardiol. 2012;154:282–6. doi: 10.1016/j.ijcard.2010.09.046[↩]
- Pietrangelo A. Hereditary hemochromatosis–a new look at an old disease. N Engl J Med. 2004 Jun 3;350(23):2383-97. doi: 10.1056/NEJMra031573[↩]
- Maurer MS, Ruberg FL. Early Diagnosis of Cardiac Amyloidosis by Carpal Tunnel Surgery: Is it All in the Wrist? J Am Coll Cardiol. 2018 Oct 23;72(17):2051-2053. doi: 10.1016/j.jacc.2018.09.003[↩]
- Porter JL, Rawla P. Hemochromatosis. [Updated 2024 Oct 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK430862[↩]
- Damy T, Maurer MS, Rapezzi C, Planté-Bordeneuve V, Karayal ON, Mundayat R, Suhr OB, Kristen AV. Clinical, ECG and echocardiographic clues to the diagnosis of TTR-related cardiomyopathy. Open Heart. 2016 Feb 8;3(1):e000289. doi: 10.1136/openhrt-2015-000289[↩][↩][↩][↩]
- Muchtar E, Blauwet LA, Gertz MA. Restrictive Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ Res. 2017 Sep 15;121(7):819-837. doi: 10.1161/CIRCRESAHA.117.310982[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Rammos A, Meladinis V, Vovas G, Patsouras D. Restrictive Cardiomyopathies: The Importance of Noninvasive Cardiac Imaging Modalities in Diagnosis and Treatment-A Systematic Review. Radiol Res Pract. 2017;2017:2874902. doi: 10.1155/2017/2874902[↩][↩]
- Kubo T., Gimeno J.R., Bahl A., Steffensen U., Steffensen M., Osman E., Thaman R., Mogensen J., Elliott P.M., Doi Y., et al. Prevalence, clinical significance, and genetic basis of hypertrophic cardiomyopathy with restrictive phenotype. J. Am. Coll. Cardiol. 2007;49:2419–2426. doi: 10.1016/j.jacc.2007.02.061[↩][↩]
- Restrictive cardiomyopathy – cardiac amyloidosis. https://radiopaedia.org/cases/restrictive-cardiomyopathy-cardiac-amyloidosis-1?lang=us[↩]
- Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997 Jan 23;336(4):267-76. doi: 10.1056/NEJM199701233360407[↩]
- Elliott P., Andersson B., Arbustini E., Bilinska Z., Cecchi F., Charron P., Dubourg O., Kuhl U., Maisch B., McKenna W.J., et al. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. Eur. Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342[↩]
- Butzner M., Leslie D.L., Cuffee Y., Hollenbeak C.S., Sciamanna C., Abraham T. Stable rates of obstructive hypertrophic cardiomyopathy in a contemporary era. Front. Cardiovasc. Med. 2021;8:765876. doi: 10.3389/fcvm.2021.765876[↩]
- Beaton A, Mocumbi AO. Diagnosis and Management of Endomyocardial Fibrosis. Cardiol Clin. 2017 Feb;35(1):87-98. doi: 10.1016/j.ccl.2016.08.005[↩]
- Towbin JA. Inherited cardiomyopathies. Circ J. 2014;78(10):2347-56. doi: 10.1253/circj.cj-14-0893[↩]
- Armanious MA, Mohammadi H, Khodor S, Oliver DE, Johnstone PA, Fradley MG. Cardiovascular effects of radiation therapy. Curr Probl Cancer. 2018 Jul;42(4):433-442. doi: 10.1016/j.currproblcancer.2018.05.008[↩]
- Mubarik A, Iqbal AM. Loeffler Endocarditis. [Updated 2024 Jan 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK534850[↩][↩][↩]
- Dregoesc MI, Iancu AC, Lazar AA, Balanescu S. Hypereosinophilic syndrome with cardiac involvement in a patient with multiple malignancies. Med Ultrason. 2018 Aug 30;20(3):399-400. doi: 10.11152/mu-1574[↩]
- Gao M, Zhang W, Zhao W, Qin L, Pei F, Zheng Y. Loeffler endocarditis as a rare cause of heart failure with preserved ejection fraction: A case report and review of literature. Medicine (Baltimore). 2018 Mar;97(11):e0079. doi: 10.1097/MD.0000000000010079[↩]
- Bhatti K, Bandlamudi M, Lopez-Mattei J. Endomyocardial Fibrosis. [Updated 2024 Oct 6]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK513293[↩]
- Allderdice C, Marcu C, Kabirdas D. Intracardiac Thrombus in Leukemia: Role of Cardiac Magnetic Resonance Imaging in Eosinophilic Myocarditis. CASE (Phila). 2018 Apr 14;2(3):114-117. doi: 10.1016/j.case.2017.12.003[↩]
- Jin X, Ma C, Liu S, Guan Z, Wang Y, Yang J. Cardiac involvements in hypereosinophilia-associated syndrome: Case reports and a little review of the literature. Echocardiography. 2017 Aug;34(8):1242-1246. doi: 10.1111/echo.13573[↩]
- Masui T, Finck S, Higgins CB. Constrictive pericarditis and restrictive cardiomyopathy: evaluation with MR imaging. Radiology. 1992 Feb;182(2):369-73. doi: 10.1148/radiology.182.2.1732952[↩]
- Cotroneo J, Sleik KM, Rene Rodriguez E, Klein AL. Hydroxychloroquine-induced restrictive cardiomyopathy. Eur J Echocardiogr. 2007 Aug;8(4):247-51. doi: 10.1016/j.euje.2006.02.002[↩]
- Okada DR, Smith J, Derakhshan A, Gowani Z, Misra S, Berger RD, Calkins H, Tandri H, Chrispin J. Ventricular Arrhythmias in Cardiac Sarcoidosis. Circulation. 2018 Sep 18;138(12):1253-1264. doi: 10.1161/CIRCULATIONAHA.118.034687[↩]
- Garcia MJ. Constrictive Pericarditis Versus Restrictive Cardiomyopathy? J Am Coll Cardiol. 2016 May 3;67(17):2061-76. doi: 10.1016/j.jacc.2016.01.076[↩][↩]
- Geske JB, Anavekar NS, Nishimura RA, Oh JK, Gersh BJ. Differentiation of Constriction and Restriction: Complex Cardiovascular Hemodynamics. J Am Coll Cardiol. 2016 Nov 29;68(21):2329-2347. doi: 10.1016/j.jacc.2016.08.050[↩]
- Pereira NL, Grogan M, Dec GW. Spectrum of Restrictive and Infiltrative Cardiomyopathies: Part 1 of a 2-Part Series. J Am Coll Cardiol. 2018 Mar 13;71(10):1130-1148. doi: 10.1016/j.jacc.2018.01.016[↩]
- Vergaro G, Aimo A, Barison A, Genovesi D, Buda G, Passino C, Emdin M. Keys to early diagnosis of cardiac amyloidosis: red flags from clinical, laboratory and imaging findings. Eur J Prev Cardiol. 2020 Nov;27(17):1806-1815. doi: 10.1177/2047487319877708[↩]
- Messroghli DR, Moon JC, Ferreira VM, Grosse-Wortmann L, He T, Kellman P, Mascherbauer J, Nezafat R, Salerno M, Schelbert EB, Taylor AJ, Thompson R, Ugander M, van Heeswijk RB, Friedrich MG. Clinical recommendations for cardiovascular magnetic resonance mapping of T1, T2, T2* and extracellular volume: A consensus statement by the Society for Cardiovascular Magnetic Resonance (SCMR) endorsed by the European Association for Cardiovascular Imaging (EACVI). J Cardiovasc Magn Reson. 2017 Oct 9;19(1):75. doi: 10.1186/s12968-017-0389-8. Erratum in: J Cardiovasc Magn Reson. 2018 Feb 7;20(1):9. doi: 10.1186/s12968-017-0408-9[↩]
- Merlo M, Gagno G, Baritussio A, Bauce B, Biagini E, Canepa M, Cipriani A, Castelletti S, Dellegrottaglie S, Guaricci AI, Imazio M, Limongelli G, Musumeci MB, Parisi V, Pica S, Pontone G, Todiere G, Torlasco C, Basso C, Sinagra G, Filardi PP, Indolfi C, Autore C, Barison A. Clinical application of CMR in cardiomyopathies: evolving concepts and techniques : A position paper of myocardial and pericardial diseases and cardiac magnetic resonance working groups of Italian society of cardiology. Heart Fail Rev. 2023 Jan;28(1):77-95. doi: 10.1007/s10741-022-10235-9[↩]
- Ryšavá R. AL amyloidosis: advances in diagnostics and treatment. Nephrol Dial Transplant. 2019 Sep 1;34(9):1460-1466. doi: 10.1093/ndt/gfy291[↩]
- Doubková M, Panovský R. Jak diagnostikovat sarkoidózu srdce? [How to diagnose cardiac sarcoidosis?]. Vnitr Lek. 2018 Fall;64(7-8):729-733. Czech.[↩]
- Baldo G, Tavares AM, Gonzalez E, Poletto E, Mayer FQ, Matte UD, Giugliani R. Progressive heart disease in mucopolysaccharidosis type I mice may be mediated by increased cathepsin B activity. Cardiovasc Pathol. 2017 Mar-Apr;27:45-50. doi: 10.1016/j.carpath.2017.01.001[↩]
- Grafft CA, Fervenza FC, Semret MH, Orloff S, Sethi S. Renal involvement in Neimann-Pick Disease. NDT Plus. 2009 Dec;2(6):448-51. doi: 10.1093/ndtplus/sfp101[↩]
- Burrow TA, Sun Y, Prada CE, Bailey L, Zhang W, Brewer A, Wu SW, Setchell KDR, Witte D, Cohen MB, Grabowski GA. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: clinical, histopathologic, and biochemical findings. Mol Genet Metab. 2015 Feb;114(2):233-241. doi: 10.1016/j.ymgme.2014.08.011[↩]
- Sweet ME, Mestroni L, Taylor MRG. Genetic Infiltrative Cardiomyopathies. Heart Fail Clin. 2018 Apr;14(2):215-224. doi: 10.1016/j.hfc.2017.12.003[↩]
- Thosani N, Younes M, Pan JJ. A heart of stone. Gastroenterology. 2013 Jul;145(1):e6-e7. doi: 10.1053/j.gastro.2013.03.026[↩]




{kind=link}